
Synthesis, Selective Cytotoxic Activity against Human Breast Cancer MCF7 Cell Line and Molecular Docking of Some Chalcone-Dihydropyrimidone Hybrids

 , , and
, , and
Abstract
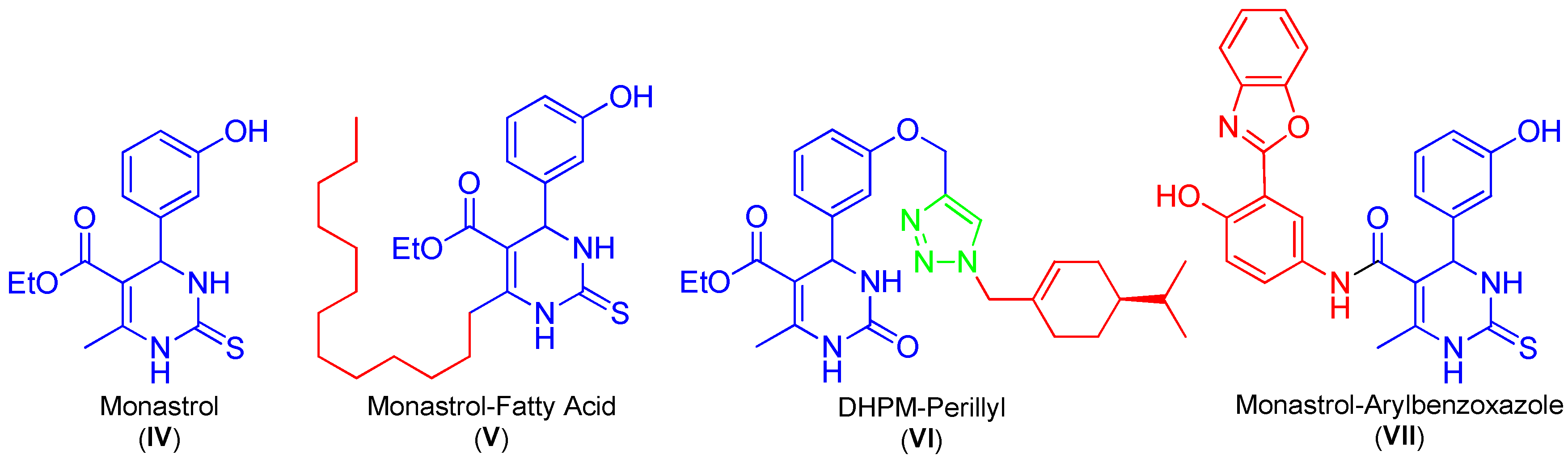
:1. Introduction
2. Results and Discussion
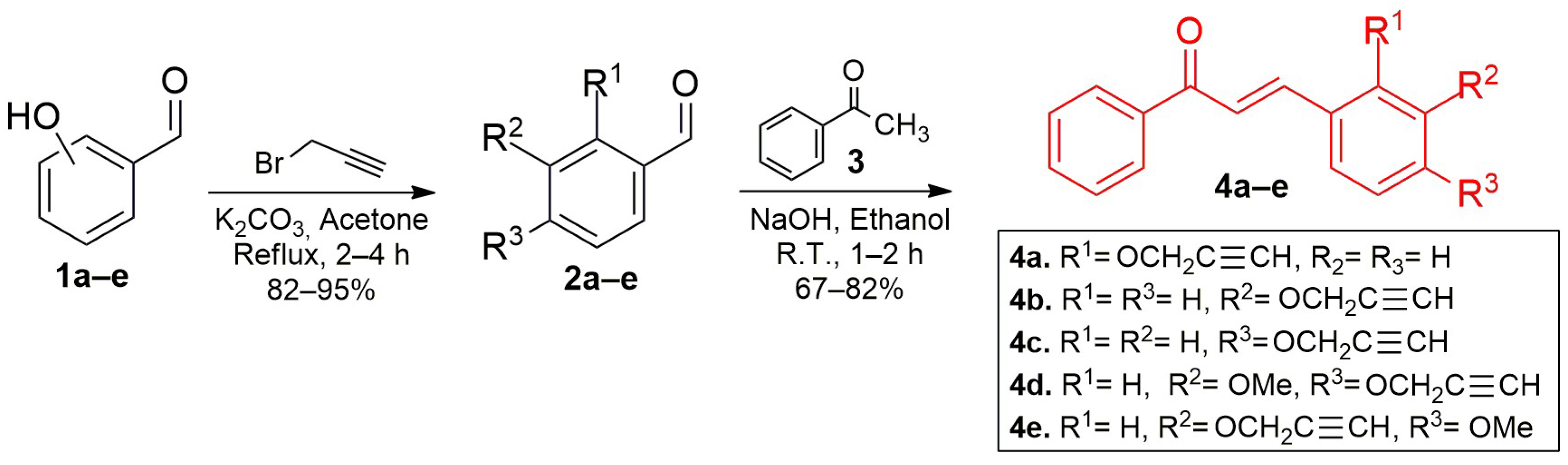
2.1. Syntehsis of Chalcone-DHPM Hybrids
2.2. Cytotoxicity of Chalcone-DHPM Hybrids
2.3. Comparative Cytotoxicity Profile of Hybrids and Their Parental Molecules
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Synthesis of Propargyloxy Benzaldehydes (2a–e) [43]
2-(Prop-2-ynil-1-oxy)benzaldehyde (2a)
3-(Prop-2-ynil-1-oxy)benzaldehyde (2b)
4-(Prop-2-ynil-1-oxy)benzaldehyde (2c)
3-Methoxy-4-(prop-2-ynil-1-oxy)benzaldehyde (2d)
4-Methoxy-3-(prop-2-ynil-1-oxy)benzaldehyde (2e)
3.1.2. General Procedure for Synthesis of Chalcones (4a–e) [44]
1-Phenyl-3-(2-prop-2-ynil-1-oxyphenyl)-(2E)-propen-1-one (4a)
1-Phenyl-3-(3-prop-2-ynil-1-oxyphenyl)-(2E)-propen-1-one (4b)
1-Phenyl-3-(4-prop-2-ynil-1-oxyphenyl)-(2E)-propen-1-one (4c)
1-Phenyl-3-(3-methoxy-4-prop-2-ynil-1-oxyphenyl)-(2E)-propen-1-one (4d)
1-Phenyl-3-(4-methoxy-3-prop-2-ynil-1-oxyphenyl)-(2E)-propen-1-one (4e)
3.1.3. General Procedure for the Synthesis of Chloro-DHPMs (7a,b) [45]
Ethyl 6-(2-chloromethyl)-4-phenyl-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (7a)
Ethyl 6-chloromethyl-4-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (7b)
3.1.4. General Procedure for the Synthesis of Azido-DHPMs (8a,b) [46]
Ethyl 6-(azidomethyl)-4-phenyl-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (8a)
Ethyl 6-(azidomethyl)-4-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (8b)
3.1.5. General Procedure for the Synthesis of Chalcone-DHPM Hybrids (9a–j) [47]
Ethyl 6-((4-((2-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-phenyl-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9a)
Ethyl 6-((4-((3-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3- triazol-1-yl)methyl)-4-phenyl-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9b)
Ethyl 6-((4-((4-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-phenyl-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9c)
Ethyl 6-((2-methoxy-4-((4-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-phenyl-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9d)
Ethyl 6-((3-methoxy-4-((3-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-phenyl-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9e)
6-((4-((2-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidin-(1H)-2-one-5 carboxylate (9f)
Ethyl 6-((4-((3-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9g)
Ethyl 6-((4-((4-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9h)
Ethyl 6-((3-methoxy-4-((4-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy)methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-(3,4,5-trimethoxyphenyl-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9i)
Ethyl 6-((3-methoxy-4-((3-(3-oxo-3-phenylprop-1-(E)-en-1-yl)phenoxy) methyl)-(1H)-1,2,3-triazol-1-yl)methyl)-4-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidin-(1H)-2-one-5-carboxylate (9j)
3.2. Cell Culture and MTT Assay
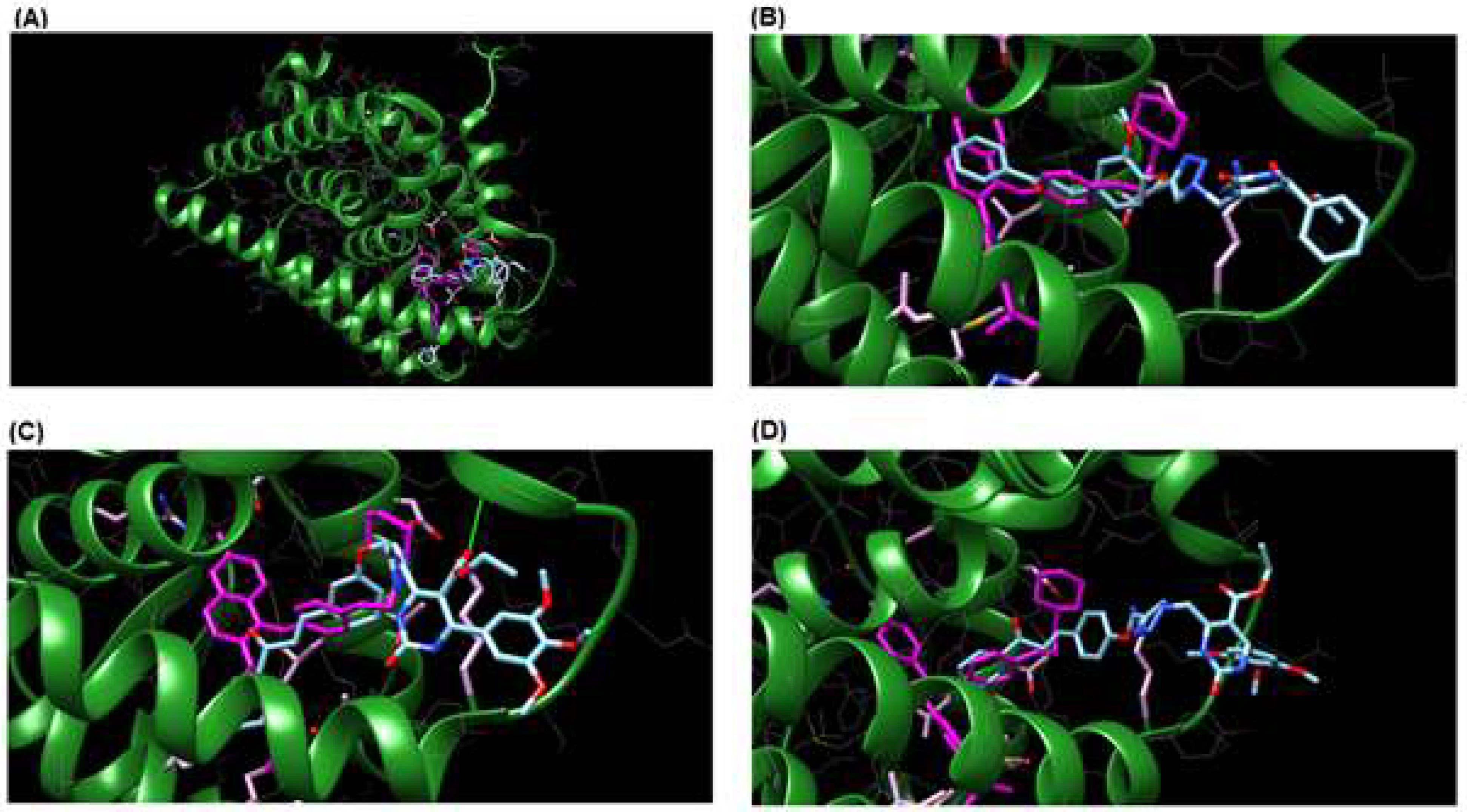
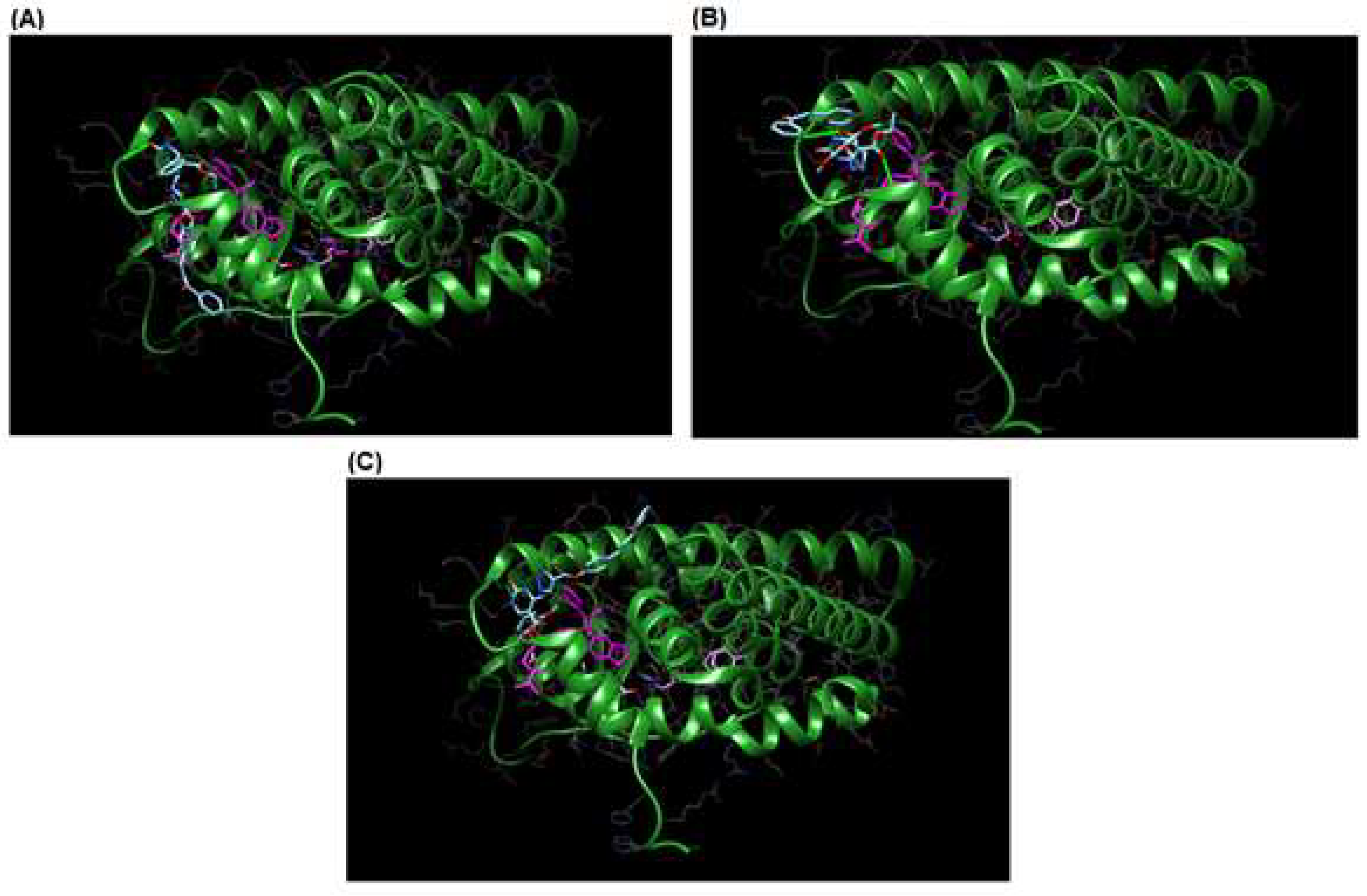
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bérubé, G. An Overview of Molecular Hybrids in Drug Discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational Approaches, Design Strategies, Structure Activity Relationship and Mechanistic Insights for Anticancer Hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Decker, M. (Ed.) Design of Hybrid Molecules for Drug Development; Elsevier: Cambridge, MA, USA, 2017. [Google Scholar]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent Progress in the Development of Synthetic Hybrids of Natural or Unnatural Bioactive Compounds for Medicinal Chemistry. Mini Rev. Med. Chem. 2010, 10, 773–793. [Google Scholar] [CrossRef]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural Product Hybrids as New Leads for Drug Discovery. Angew. Chem. Int. Ed. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Vendrusculo, V.; de Souza, V.P.; Fontoura, L.A.M.; D’Oca, M.G.M.; Banzato, T.P.; Monteiro, P.A.; Pilli, R.A.; de Carvalho, J.E.; Russowsky, D. Synthesis of Novel Perillyl-Dihydropyrimidinone Hybrids Designed for Antiproliferative Activity. Med. Chem. Commun. 2018, 9, 1553–1564. [Google Scholar] [CrossRef]
- Sharma, M.; Chauhan, K.; Shivahare, R.; Vishwakarma, P.; Suthar, M.K.; Sharma, A.; Gupta, S.; Saxena, J.K.; Lal, J.; Chandra, P.; et al. Discovery of a New Class of Natural Product-Inspired Quinazolinone Hybrid as Potent Antileishmanial agents. J. Med. Chem. 2013, 56, 4374–4392. [Google Scholar] [CrossRef]
- Camps, P.; Achab, R.E.; Morral, J.; Muñoz-Torrero, D.; Badia, A.; Baños, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; Luque, F.J. New Tacrine-Huperzine A Hybrids (Huprines): Highly Potent Tight-Binding Acetylcholinesterase Inhibitors of Interest for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2000, 43, 4657–4666. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A mini review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Anand, A.; Kumar, K.; Kumar, V. Recent developments in biological aspects of chalcones: The odyssey continues. Expert Opin. Drug Discov. 2019, 14, 249–288. [Google Scholar] [CrossRef] [PubMed]
- Salum, L.B.; Altei, W.F.; Chiaradia, L.D.; Cordeiro, M.N.S.; Canevarolo, R.R.; Melo, C.P.S.; Winter, E.; Mattei, B.; Daghestani, H.N.; Santos-Silva, M.C.; et al. Cytotoxic 3,4,5-Trimethoxychalcones as Mitotic Arresters and Cell Migration Inhibitors. Eur. J. Med. Chem. 2013, 63, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankaraiah, N.; Nekkanti, S.; Brahma, U.R.; Praveen Kumar, N.; Deshpande, N.; Prasanna, D.; Senwar, K.R.; Jaya Lakshmi, U. Synthesis of Different Heterocycles-Linked Chalcone Conjugates as Cytotoxic Agents and Tubulin Polymerization Inhibitors. Bioorgan. Med. Chem. 2017, 25, 4805–4816. [Google Scholar] [CrossRef]
- Wei, H.; Ruan, J.; Zhang, X. Coumarin-chalcone hybrids: Promising agents with diverse pharmacological properties. RSC Adv. 2016, 6, 10846–10860. [Google Scholar] [CrossRef]
- Mao, Z.; Zheng, X.; Qi, Y.; Zhang, M.; Huang, Y.; Wan, C.; Rao, G. Synthesis and biological evaluation of novel hybrid compounds between chalcone and piperazine as potential antitumor agents. RSC Adv. 2016, 6, 7723–7727. [Google Scholar] [CrossRef]
- Stanojković, T.; Marković, V.; Matić, I.Z.; Mladenović, M.P.; Petrovića, N.; Krivokuća, A.; Petković, M.; Joksović, M.D. Highly selective anthraquinone-chalcone hybrids as potential antileukemia agents. Bioorgan. Med. Chem. Lett. 2018, 28, 2593–2598. [Google Scholar] [CrossRef]
- Park, S.; Kim, E.H.; Kim, J.; Kim, S.H.; Kim, I. Biological evaluation of indolizine-chalcone hybrids as new anticancer agents. Eur. J. Med. Chem. 2018, 144, 435–443. [Google Scholar] [CrossRef]
- Thiriveedhi, A.; Nadh, R.V.; Srinivasu, N.; Kaushal, K. Novel Hybrid Molecules of Quinazoline Chalcone Derivatives: Synthesis and Study of In Vitro Cytotoxic Activities. Lett. Drug Des. Discov. 2018, 15, 757–765. [Google Scholar] [CrossRef]
- Mass, E.B.; Duarte, G.V.; Russowsky, D. The Quinazoline-Chalcone and Quinazolinone-Chalcone Hybrids: A Promising Combination for Biological Activity. Mini Rev. Med. Chem. 2021, 21, 186–203. [Google Scholar] [CrossRef] [PubMed]
- Jernei, T.; Duró, C.; Dembo, A.; Lajkó, E.; Takács, A.; Kohidai, L.; Schlosser, G.; Csámpai, A. Synthesis, Structure and In Vitro Cytotoxic Activity of Novel Cinchona-Chalcone Hybrids with 1,4-Disubstituted and 1,5-Disubstituted 1,2,3-Triazole Linkers. Molecules 2019, 24, 4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, S.H.; El-Hafeez, A.A.A.; Shoman, M.E.; Montano, M.M.; Hassan, H.A. New quinoline-chalcone hybrids as anti-cancer agents: Design, synthesis, and evaluations of cytotoxicity and PI3K inhibitory activity. Bioorgan. Chem. 2019, 82, 360–377. [Google Scholar] [CrossRef] [PubMed]
- Chouiter, M.I.; Boulebd, H.; Pereira, D.M.; Valentao, P.; Andrade, P.B.; Belfaitah, A.; Silva, A.M.S. New chalcone-type compounds and 2-pyrazoline derivatives: Synthesis and caspase-dependent anticancer activity. Future Med. Chem. 2020, 12, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Djemoui, A.; Naouri, A.; Ouahrani, M.R.; Djemoui, D.; Lahcene, S.; Lahrech, M.B.; Boukenna, L.; Albuquerque, H.M.T.; Saher, L.; Rocha, D.H.A.; et al. A step-by-step synthesis of triazole-benzimidazole-chalcone hybrids: Anticancer activity in human cells. J. Mol. Struct. 2020, 1204, 127487. [Google Scholar] [CrossRef]
- Bukhari, S.N.A.; Jasamai, M.; Jantan, I.; Ahmad, W. Review of Methods and Various Catalysts Used for Chalcone Synthesis. Mini Rev. Org. Chem. 2013, 10, 73–83. [Google Scholar] [CrossRef]
- Qin, H.L.; Zhang, Z.W.; Lekkala, R.; Alsulami, H.; Rakesh, K.P. Chalcone hybrids as privileged scaffolds in antimalarial drug discovery: A key review. Eur. J. Med. Chem. 2020, 193, 112215. [Google Scholar] [CrossRef]
- Gao, F.; Huang, G.; Xiao, J. Chalcone hybrids as potential anticancer agents: Current development, mechanism of action, and structure-activity relationship. Med. Res. Rev. 2020, 40, 2049–2084. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide–alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- Neumann, S.; Biewend, M.; Rana, S.; Binder, W.H. The CuAAC: Principles, homogeneous and heterogeneous catalysts, and novel developments and applications. Macromol. Rapid Commun. 2019, 41, 1900359. [Google Scholar] [CrossRef]
- Lal, K.; Yadav, P.; Kumar, A.; Kumar, A.; Paul, A.K. Design, synthesis, characterization, antimicrobial evaluation and molecular modeling studies of some dehydroacetic acid-chalcone-1,2,3-triazole hybrids. Bioorgan. Chem. 2018, 77, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Raj, R.; Kumar, V.; Mahajan, M.P.; Bedi, P.M.S.; Kaur, T.; Saxena, A.K. 1,2,3-Triazole tethered β-lactam-chalcone bifunctional hybrids: Synthesis and anticancer evaluation. Eur. J. Med. Chem. 2012, 47, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Guantai, E.M.; Ncokazi, K.; Egan, T.J.; Gut, J.; Rosenthal, P.J.; Smith, P.J.; Chibale, K. Design, synthesis and in vitro antimalarial evaluation of triazole-linked chalcone and dienone hybrid compounds. Bioorgan. Med. Chem. 2010, 18, 8243–8256. [Google Scholar] [CrossRef] [PubMed]
- Mayer, T.U.; Kapoor, T.M.; Haggarty, S.J.; King, R.W.; Schreiber, S.L.; Mitchison, T.J. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999, 286, 971–974. [Google Scholar] [CrossRef] [Green Version]
- Cochran, J.C.; Gatial, J.E.; Kapoor, T.M.; Gilbert, S.P. Monastrol inhibition of the mitotic kinesin Eg5. J. Biol. Chem. 2005, 280, 12658–12667. [Google Scholar] [CrossRef] [Green Version]
- Matos, L.H.S.; Masson, F.T.; Simeoni, L.A.; Homem-de-Mello, M. Biological Activity of Dihydropyrimidinone (DHPM) Derivatives: A Systematic Review. Eur. J. Med. Chem. 2018, 143, 1779–1789. [Google Scholar] [CrossRef]
- Kaur, R.; Chaudhary, S.; Kumar, K.; Gupta, M.K.; Rawal, R.K. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A Review. Eur. J. Med. Chem. 2017, 132, 108–134. [Google Scholar] [CrossRef]
- de Fátima, Â.; Braga, T.C.; Neto, L.S.; Terra, B.S.; Oliveira, B.G.F.; da Silva, D.L.; Modolo, L.V. A Mini-Review on Biginelli adducts with notable pharmacological properties. J. Adv. Res. 2015, 6, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, F.S.; Oliveira, P.M.; Farias, L.M.; Brinkerhoff, R.C.; Sobrinho, R.C.M.A.; Treptow, T.M.; D’Oca, C.R.M.; Marinho, M.A.G.; Hort, M.A.; Horn, A.P.; et al. Synthesis and antitumoral activity of novel hybrid monastrol-fatty acids against glioma cells. Med. Chem. Commun. 2018, 9, 1282–1288. [Google Scholar] [CrossRef]
- Souza, V.P.; Santos, F.S.; Rodembusch, F.S.; Braga, C.B.; Ornelas, C.; Pilli, R.A.; Russowsky, D. Hybrid 3,4-dihydropyrimidin-2-(thi)ones as dual-functional bioactive molecules: Fluorescent probes and cytotoxic agents to cancer cells. New J. Chem. 2020, 44, 12440. [Google Scholar] [CrossRef]
- Malik, M.S.; Ahmed, S.A.; Althagafi, I.I.; Ansari, M.A.; Kama, A. Application of triazoles as bioisosteres and linkers in the development of microtubule targeting agents. RSC Med. Chem. 2020, 11, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Hau, S.C.K.; Cheng, P.S.; Mak, T.C.W. Assembly of organosilver (I) frameworks with terminal ethynide and ethenyl groups on separate pendent arms attached to an aromatic ring. Polyhedron 2013, 52, 992–1008. [Google Scholar] [CrossRef]
- Chang, M.Y.; Tai, H.Y.; Chen, Y.L.; Hsu, R.T. Synthesis of 1,3-Diaryl-1H-Benzo[g]Indazoles. Tetrahedron 2012, 68, 7941–7948. [Google Scholar] [CrossRef]
- Lebed, P.S.; Kos, P.O.; Polovinko, V.V.; Tolmachev, A.A.; Vovk, M.V. Synthesis of new polyfunctional 5,6,7,8-tetrahydroimidazo-[1,5-c]pyrimidin-5-ones by the aza-Wittig reaction followed by intramolecular cyclization and 1,3-prototropic Shift. Russ. J. Org. Chem. 2009, 45, 921–927. [Google Scholar] [CrossRef]
- Chen, J.; Fu, X.; Zhou, L.; Zhang, J.; Qi, X.; Cao, X. A convergent route for the total synthesis of Malyngamides O, P, Q and R. J. Org. Chem. 2009, 74, 4149–4157. [Google Scholar] [CrossRef]
- Moro, A.V.; Ferreira, P.C.; Migowski, P.; Rodembusch, F.S.; Dupont, J.; Lüdtke, D.S. Synthesis and photophysical properties of fluorescent 2,1,3-benzothiadiazole-triazole-linked glycoconjugates: Selective chemosensors for Ni (II). Tetrahedron 2013, 69, 201–206. [Google Scholar] [CrossRef]
- Priti, K.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 6, 469–471. [Google Scholar]
- Afonso de Lima, C.; de Souza Bueno, I.L.; Nunes Siqueira Vasconcelos, S.; Sciani, J.M.; Ruiz, A.L.T.G.; Foglio, M.A.; Carvalho, J.E.D.; Barbarini Longato, G. Reversal of Ovarian Cancer Cell Lines Multidrug Resistance Phenotype by the Association of Apiole with Chemotherapies. Pharmaceuticals 2020, 13, 327. [Google Scholar] [CrossRef]
- Suffness, M.; Pezzuto, J.M. Methods in Plant Biochemistry: Assays for Bioactivity; Hostettmann, K., Ed.; Academic Press: London, UK, 1990; Volume 6, pp. 71–133. [Google Scholar]
- Zhou, W.; Pan, T.; Cui, H.; Zhao, Z.; Chu, P.K.; Yu, X.-F. Black Phosphorus: Bioactive Nanomaterials with Inherent and Selective Chemotherapeutic Effects. Angew. Chem. Int. Ed. 2019, 58, 769–774. [Google Scholar] [CrossRef]
- Brooks, S.C.; Locke, E.R.; Soule, H.D. Estrogen Receptor in a Human Cell Line (MCF-7) from Breast Carcinoma. J. Biol. Chem. 1973, 17, 6251–6253. [Google Scholar] [CrossRef]
- Penot, G.; Peron, C.L.; Merot, Y.; Grimaud-Fanouillère, E.; Ferrière, F.; Boujrad, B.; Kah, O.; Saligaut, C.; Ducouret, B.; Métivier, R.; et al. The Human Estrogen Receptor-α Isoform hERα46 Antagonizes the Proliferative Influence of hERα66 in MCF7 Breast Cancer Cells. Endocrinology 2005, 12, 5474–5484. [Google Scholar] [CrossRef] [PubMed]
- Mota, A.D.; Evangelista, A.F.; Macedo, T.; Oliveira, R.; Scapulatempo Neto, C.; Vieira, R.A.; Marques, M.M. Molecular characterization of breast cancer cell lines by clinical immunohistochemical markers. Oncol. Lett. 2017, 13, 4708–4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hans, R.H.; Guantai, E.M.; Lategan, C.; Smith, P.J.; Wan, B.; Franzblau, S.G.; Gut, J.; Rosenthal, P.J.; Chibale, K. Synthesis, Antimalarial and Antitubercular Activity of Acetylenic Chalcones. Bioorgan. Med. Chem. Lett. 2009, 20, 942–944. [Google Scholar] [CrossRef]
- Hussaini, S.M.A.; Yedla, P.; Babu, K.S.; Shaik, T.B.; Chityal, G.K.; Kamal, A. Synthesis and Biological Evaluation of 1,2,3-Triazole Tethered Pyrazoline and Chalcone Derivatives. Chem. Biol. Drug Des. 2016, 88, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Laia, F.M.R.; Pinho, E.; Melo, T.M.V.D. Synthesis and reactivity of aziridines with internal dipolarophiles: An approach to 1,4-Dihydrochromeno-[4,3-b]pyrroles and 3-Methylenechromano-[4,3-b]pyrroles. Synthesis 2015, 47, 2781–2790. [Google Scholar]
- Singh, G.; Singh, J.; Mangat, S.S.; Arora, A. Synthetic approach towards ‘Click’ modified chalcone based organo-triethoxysilanes, UV-Vis Study. RSC Adv. 2014, 4, 60853–60865. [Google Scholar] [CrossRef]
- Niu, C.; Li, G.; Tuerxuntayi, A.; Aisa, H.A. Synthesis and bioactivity of new chalcone derivatives as potential tyrosinase activator based on the Click chemistry. Chin. J. Chem. 2015, 33, 486–494. [Google Scholar] [CrossRef]
- Sharghi, H.; Jokar, M. Al2O3/MeSO3H: A novel and recyclable catalyst for one-pot synthesis of 3,4-Dihydropyrimidinones or their sulfur derivatives in Biginelli condensation. Synth. Commun. 2009, 39, 958–979. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Franco, Y.E.M.; Okubo, M.Y.; Torre, A.D.; Paiva, P.P.; Rosa, M.N.; Silva, V.A.O.; Reis, R.M.; Ruiz, A.L.T.G.; Imamura, P.M.; de Carvalho, J.E.; et al. Coronarin D Induces Apoptotic Cell Death and Cell Cycle Arrest in Human Glioblastoma Cell Line. Molecules 2019, 24, 4498. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Hybrid Compounds | H-Triazole (ppm) | Yield (%) a | |
|---|---|---|---|---|
| 1 | 9a | 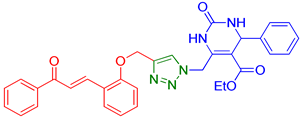 | 9.00 | 75 |
| 2 | 9b |  | 8.35 | 70 |
| 3 | 9c | 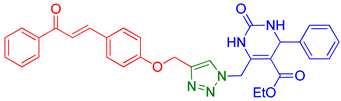 | 8.43 | 78 |
| 4 | 9d | 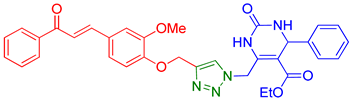 | 8.48 | 90 |
| 5 | 9e |  | 8.39 | 75 |
| 6 | 9f |  | 9.39 | 72 |
| 7 | 9g |  | 8.42 | 70 |
| 8 | 9h | 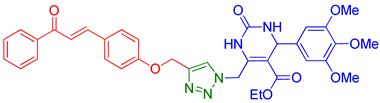 | 8.56 | 62 |
| 9 | 9i | 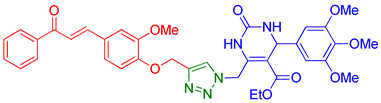 | 8.49 | 90 |
| 10 | 9j |  | 8.43 | 85 |
| Hybrid | U-251 | MCF-7 | NCI/ADR-RES | OVCAR-3 | HT-29 | HaCaT |
|---|---|---|---|---|---|---|
| 9a | 27.0 ± 11.0 | 6.2 ± 3.4 | 6.4 ± 3.4 | 9.9 ± 3.5 | 18.6 ± 8.7 | 6.7 ± 3.0 |
| 9b | 26.1 ± 8.2 | 5.3 ± 2.7 | 14.5 ± 5.5 | 9.4 ± 3.4 | 43.8 ± 12.8 | 22.0 ± 3.9 |
| 9c | 65.1 ± 11.0 | 11.5 ± 5.1 | 54.8 ± 9.6 | 70.6 ± 12.1 | 134.1 ± 33.7 | 22.0 ± 3.9 |
| 9d | 74.6 ± 17.5 | 4.7 ± 2.5 | 89.8 ± 12.1 | 109.7 ± 31.8 | 109.7 ± 46.5 | 17.5 ± 2.3 |
| 9e | 24.6 ± 6.6 | 12.3 ± 3.4 | 26.8 ± 7.2 | 15.3 ± 3.4 | 41.1 ± 7.9 | 12.0 ± 2.2 |
| 9f | 10.9 ± 4.1 | 7.5 ± 2.7 | 40.4 ± 11.2 | 12.5 ± 4.7 | 25.1 ± 12.5 | 6.4 ± 3.0 |
| 9g | 13.3 ± 3.8 | 4.9 ± 1.4 | 39.6 ± 7.9 | 17.9 ± 5.8 | 93.9 ± 29.8 | 15.1 ± 4.1 |
| 9h | 70.4 ± 9.5 | 5.8 ± 2.6 | >153.0 | 36.4 ± 7.2 | >153.0 | 100.7 ± 13.0 |
| 9i | 104.1 ± 33.2 | 5.3 ± 2.5 | >146.3 | 23.5 ± 6.0 | 103.4 ± 18.1 | 34.8 ± 4.8 |
| 9j | 128.7 ± 35.5 | 14.6 ± 4.5 | >146.3 | 88.9 ± 11.0 | >146.3 | 69.3 ± 7.9 |
| DOXO | >18.4 | 0.4 ± 0.0 | 22.6 ± 3.9 | 11.6 ± 5.1 | >18.4 | 1.6 ± 0.0 |
| Cell Lines | Chalcone | DHPM | Hybrid | Selectivity Index (SI) |
|---|---|---|---|---|
| 4d | 8a | 9d (4d + 8a) | 9d | |
| U-251 | 251.51 | >332 | 74.62 | 0.23 |
| MCF-7 | >342 | >332 | 4.72 | 3.71 |
| NCI/ADR-RES | >342 | >332 | 89.79 | 0.19 |
| HaCaT | 97.05 | >332 | 17.52 | - |
| 4b | 8b | 9g (4b + 8b) | 9g | |
| U-251 | 24.47 | >255 | 13.30 | 1.14 |
| MCF-7 | 80.33 | >255 | 4.89 | 3.10 |
| NCI/ADR-RES | >381 | >255 | 39.62 | 0.38 |
| HaCaT | 23.33 | >255 | 15.14 | - |
| 4c | 8b | 9h (4c + 8b) | 9h | |
| U-251 | 126.50 | >255 | 70.37 | 1.43 |
| MCF-7 | >381 | >255 | 5.81 | 17.33 |
| NCI/ADR-RES | >381 | >255 | >153 | - |
| HaCaT | 37.59 | >255 | 100.7 | - |
| Hybrid | Binding Energy (kcal/mol) | Binding Interaction | Bond Lenght (Å) |
|---|---|---|---|
| 9d | −10.32 | Leu346 | 6.96 |
| Thr347a | 3.93 | ||
| Asp351a | 3.29 | ||
| Glu353a | 8.27 | ||
| 9g | −12.02 | Leu346 | 6.33 |
| Thr347a | 4.05 | ||
| Asp351a | 3.68 | ||
| Asn532 | 2.36 | ||
| Leu536 | 2.50 | ||
| 9h | −7.52 | Leu346 | 4.86 |
| Thr347a | 5.35 | ||
| Asp351a | 4.27 |
| Hybrid | Binding Energy (kcal/mol) | Binding Interaction | Bond Lenght (Å) |
|---|---|---|---|
| 9d | −9.38 | Cys530 | 2.38 |
| Ser537 | 9.88 | ||
| Asp351 | 4.46 | ||
| 9g | −7.85 | Cys530 | 2.59 |
| Ser537 | 7.26 | ||
| Leu536 | 2.36 | ||
| 9h | −8.51 | Cys530 | 4.24 |
| Ser537 | 7.01 | ||
| Leu536 | 7.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mass, E.B.; de Lima, C.A.; D’Oca, M.G.M.; Sciani, J.M.; Longato, G.B.; Russowsky, D. Synthesis, Selective Cytotoxic Activity against Human Breast Cancer MCF7 Cell Line and Molecular Docking of Some Chalcone-Dihydropyrimidone Hybrids. Drugs Drug Candidates 2022, 1, 3-21. https://doi.org/10.3390/ddc1010002
Mass EB, de Lima CA, D’Oca MGM, Sciani JM, Longato GB, Russowsky D. Synthesis, Selective Cytotoxic Activity against Human Breast Cancer MCF7 Cell Line and Molecular Docking of Some Chalcone-Dihydropyrimidone Hybrids. Drugs and Drug Candidates. 2022; 1(1):3-21. https://doi.org/10.3390/ddc1010002
Chicago/Turabian StyleMass, Eduardo B., Carolina A. de Lima, Marcelo G. M. D’Oca, Juliana M. Sciani, Giovanna B. Longato, and Dennis Russowsky. 2022. "Synthesis, Selective Cytotoxic Activity against Human Breast Cancer MCF7 Cell Line and Molecular Docking of Some Chalcone-Dihydropyrimidone Hybrids" Drugs and Drug Candidates 1, no. 1: 3-21. https://doi.org/10.3390/ddc1010002