
Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia

 ,
,  ,
,  , and
, and 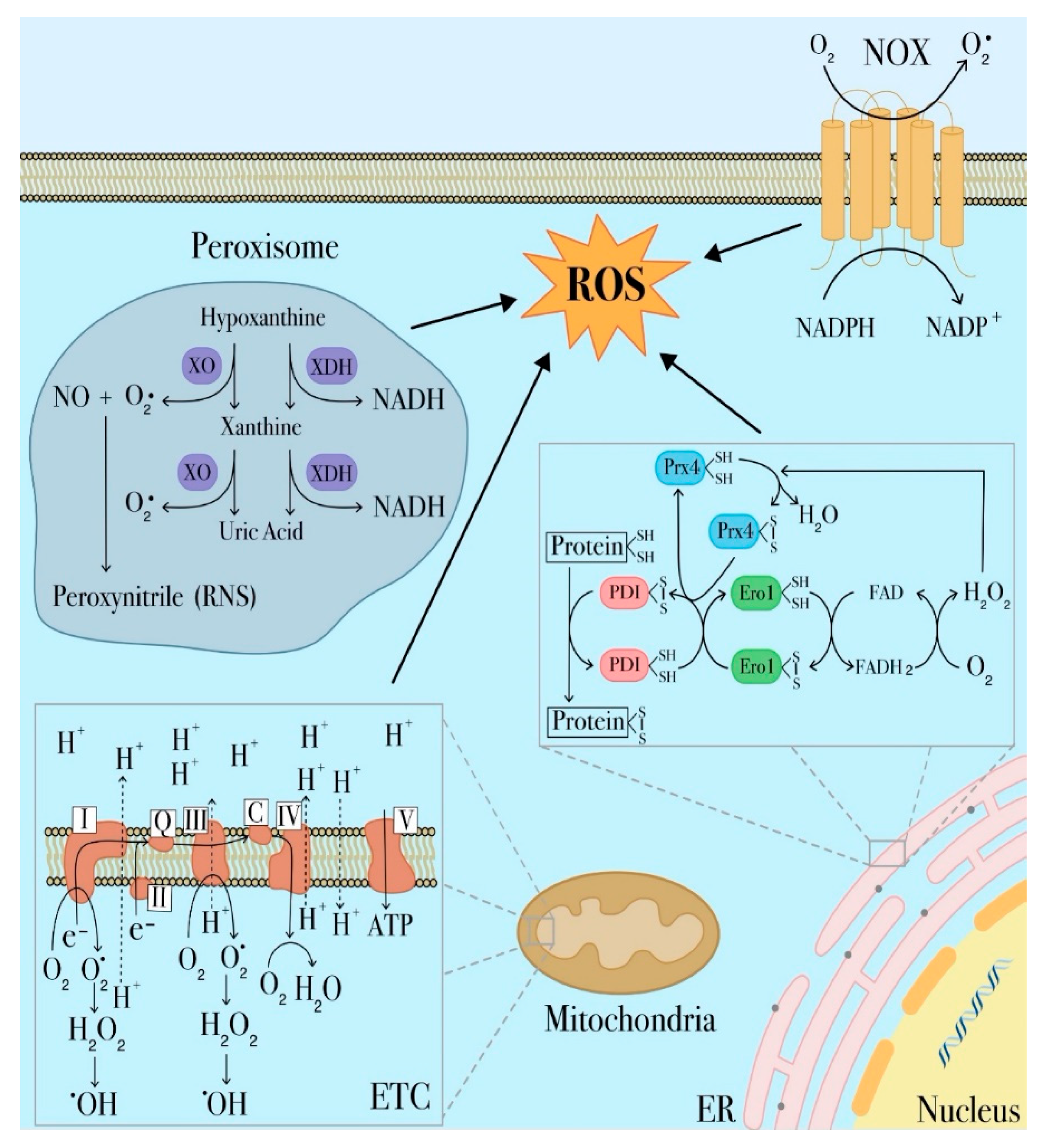{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Energy Metabolism and Cell Redox State in Myeloid Stem Cells
3. ROS Generation and Antioxidant Defense Systems in Myeloid Leukemia Cells
3.1. Sources of ROS Production
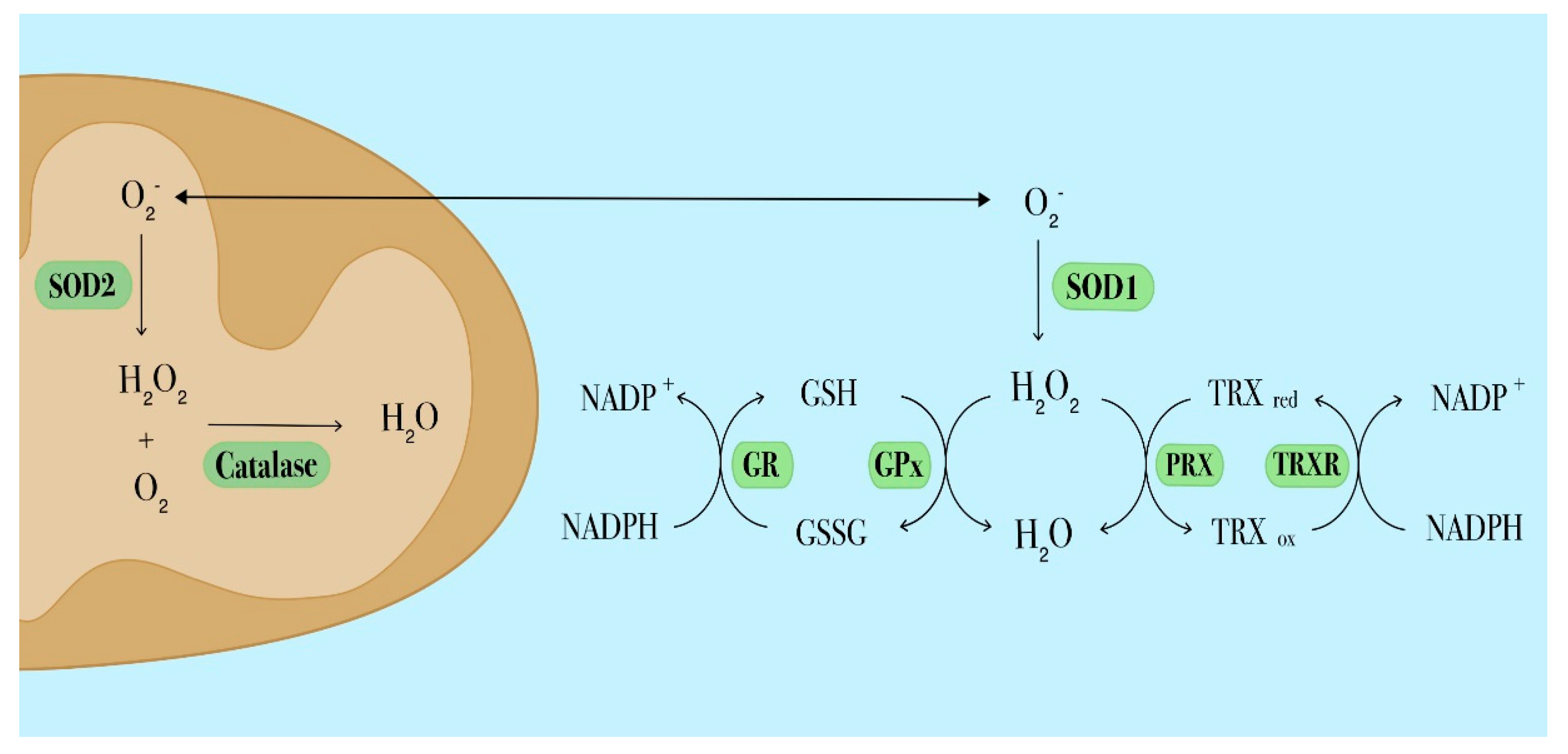
3.2. Antioxidant Defences
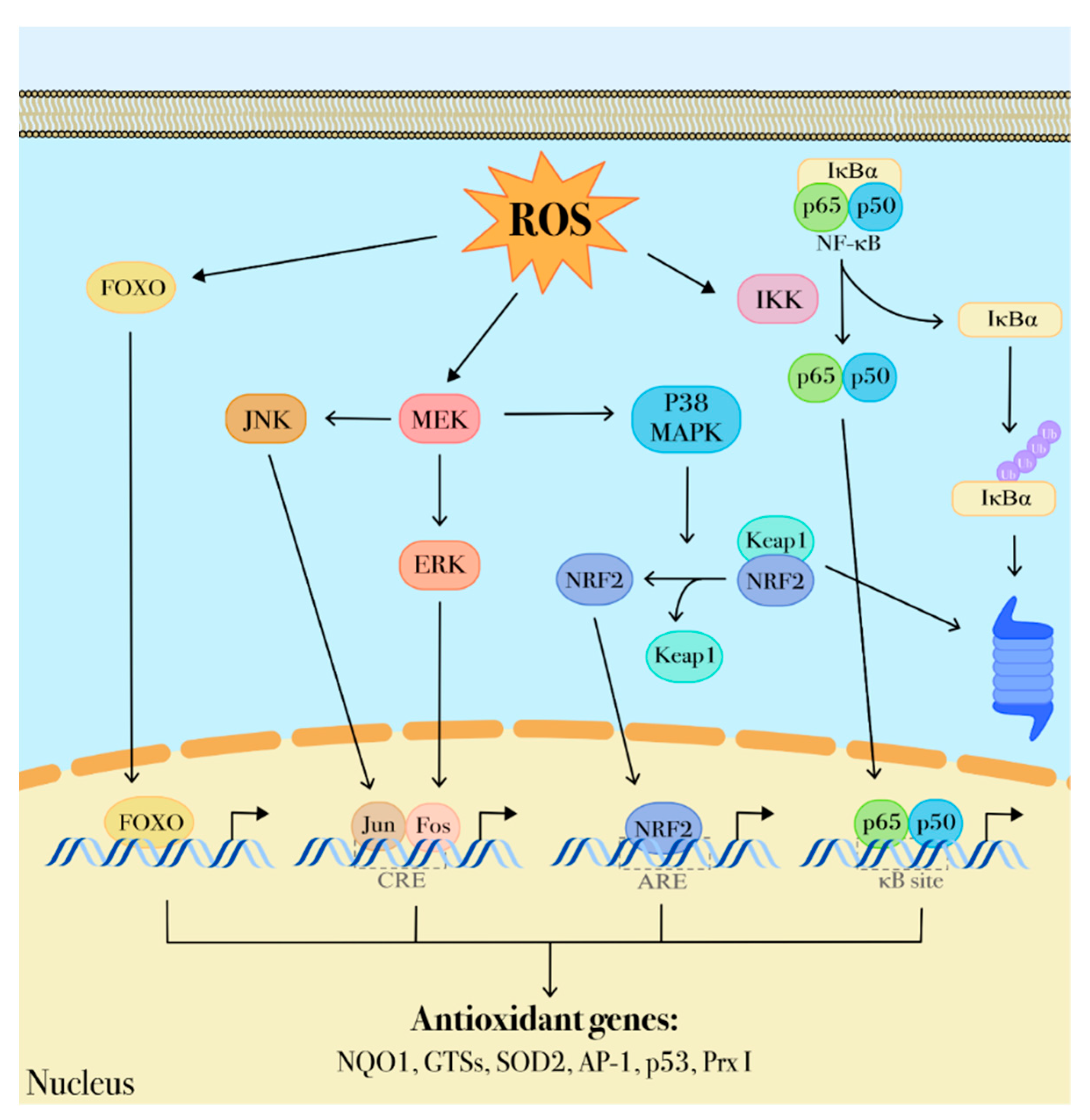
4. ROS-Mediated Signaling Pathways Involved in the Leukemogenic Process
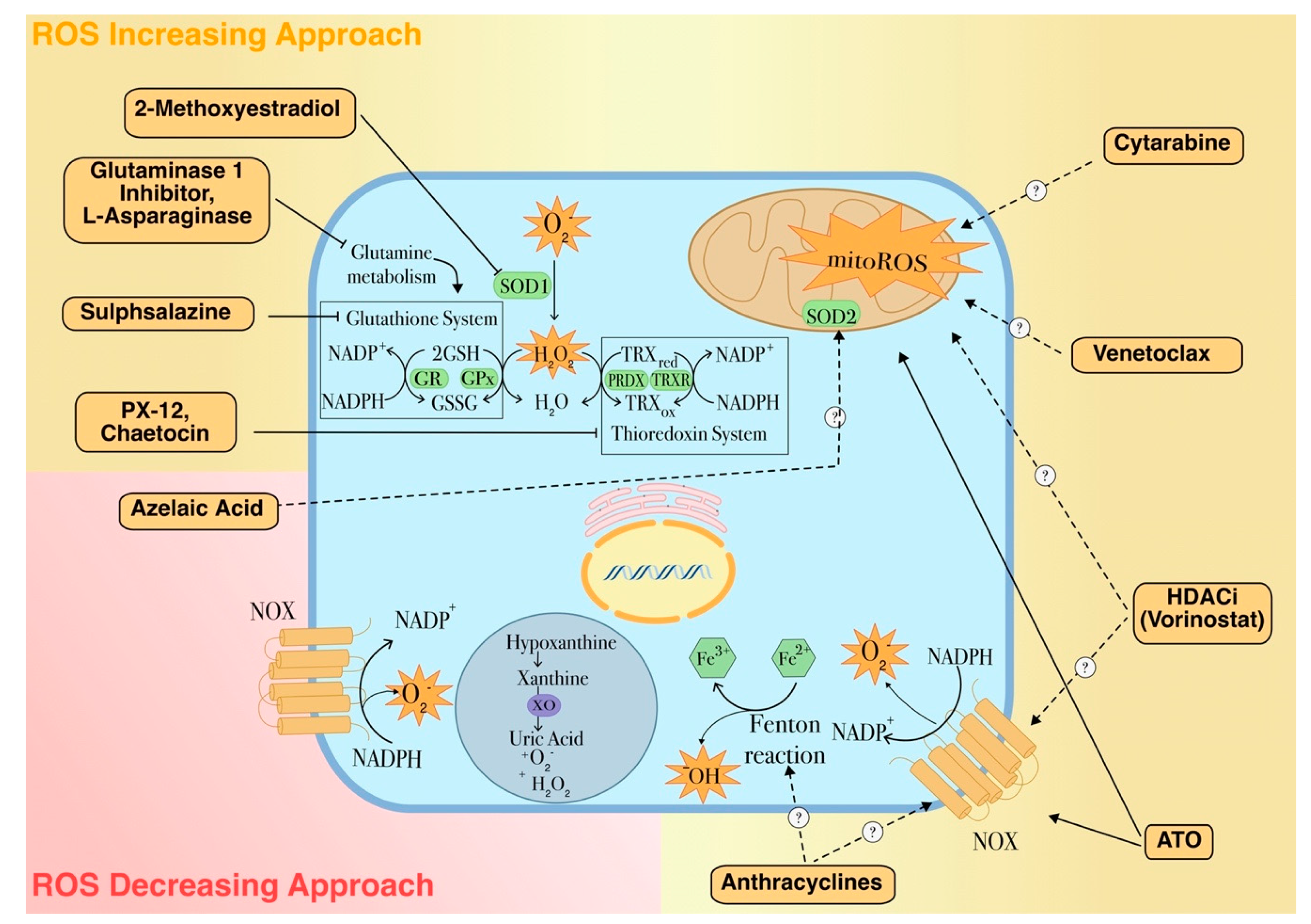
5. ROS-Based Therapies in Myeloid Leukemia Treatment
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| ARE | Antioxidant responsive elements |
| ATO | Arsenic trioxide |
| CLL | Chronic lymphocytic leukemia |
| CML | Chronic myeloid leukemia |
| CYP | Cytochrome P450 |
| ER | Endoplasmic reticulum |
| Ero1 | Endoplasmic reticulum oxidoreductin-1 |
| ETC | Electron transport chain |
| FOXO | Forkhead O box |
| GCL | Glutamate-cysteine ligase |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| GST | Glutathione S-transferases |
| HDACs | Histone deacetylases |
| HIFs | Hypoxia inducible factors |
| HO-1 | Heme oxygenase- 1 |
| HSCs | Hematopoietic stem cells |
| Keap1 | Helch-like ECH-associated protein |
| LNKHSCs | Lymphocyte adaptor proteinHematopoietic stem cells |
| LSCs | Leukemic stem cells |
| MPPs | Multi-potent progenitors |
| mTOR | Mammalian target of rapamycin |
| NAC | N-acetylcysteine |
| NF-κB | Nuclear factor-κB |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NOX | NADPH oxidase |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor (erythroid-derived)-like 2 |
| PDI | Protein disulfide isomerases |
| PTPs | Protein tyrosin phosphatases |
| PX-12 | 1-methylpropyl 2-imidazolyl disulfide |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| Tkr1 | Thioredoxin reductase 1 |
| Trx | Thioredoxin |
| UPR | Unfolded protein response |
| XDH | Xanthine dehydrogenase |
| XO | Xanthine oxidase |
| XOR | Xanthine oxidoreductase |
References
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Lapidus, R.G.; Ferraris, D.; Emadi, A. Analysis of the Mechanisms of Action of Naphthoquinone-Based Anti-Acute Myeloid Leukemia Chemotherapeutics. Molecules 2019, 24, 3121. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative Stress: Eustress and Distress in Redox Homeostasis. In Stress: Physiology, Biochemistry, and Pathology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 153–163. ISBN 978-0-12-813146-6. [Google Scholar]
- Aggarwal, V.; Tuli, H.; Varol, A.; Thakral, F.; Yerer, M.; Sak, K.; Varol, M.; Jain, A.; Khan, M.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Levine, A.S.; Sun, L.; Tan, R.; Gao, Y.; Yang, L.; Chen, H.; Teng, Y.; Lan, L. The Oxidative DNA Damage Response: A Review of Research Undertaken with Tsinghua and Xiangya Students at the University of Pittsburgh. Sci. China Life Sci. 2017, 60, 1077–1080. [Google Scholar] [CrossRef]
- Grove, C.S.; Vassiliou, G.S. Acute Myeloid Leukaemia: A Paradigm for the Clonal Evolution of Cancer? Dis. Model. Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef]
- Chen, K.T.J.; Gilabert-Oriol, R.; Bally, M.B.; Leung, A.W.Y. Recent Treatment Advances and the Role of Nanotechnology, Combination Products, and Immunotherapy in Changing the Therapeutic Landscape of Acute Myeloid Leukemia. Pharm. Res. 2019, 36, 125. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.C. Genetic Abnormalities and Challenges in the Treatment of Acute Myeloid Leukemia. Genes Cancer 2011, 2, 95–107. [Google Scholar] [CrossRef]
- Mattes, K.; Vellenga, E.; Schepers, H. Differential Redox-Regulation and Mitochondrial Dynamics in Normal and Leukemic Hematopoietic Stem Cells: A Potential Window for Leukemia Therapy. Crit. Rev. Oncol. Hematol. 2019, 144, 102814. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, D.; Guo, F.; Xuan, C. Mitochondrial Membrane Potential and Reactive Oxygen Species in Cancer Stem Cells. Fam. Cancer 2015, 14, 19–23. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive Oxygen Species in Cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 1–17. [Google Scholar] [CrossRef]
- Kaweme, N.M.; Zhou, S.; Changwe, G.J.; Zhou, F. The Significant Role of Redox System in Myeloid Leukemia: From Pathogenesis to Therapeutic Applications. Biomark. Res. 2020, 8. [Google Scholar] [CrossRef]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive Oxygen Species in Haematopoiesis: Leukaemic Cells Take a Walk on the Wild Side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fang, H.; Wang, K. Reactive Oxygen Species in Eradicating Acute Myeloid Leukemic Stem Cells. Stem Cell Investig. 2014, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K.; Schnorr, P.J.; Pang, W.W.; Chao, M.P.; Chhabra, A.; Seita, J.; Feng, M.; Weissman, I.L. Myeloid Cell Origins, Differentiation, and Clinical Implications. Microbiol. Spectr. 2016, 4, 857–875. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, Y.; Luo, X.; Hu, Q. Oxidative Resistance of Leukemic Stem Cells and Oxidative Damage to Hematopoietic Stem Cells under Pro-Oxidative Therapy. Cell Death Dis. 2020, 11, 291. [Google Scholar] [CrossRef]
- Chao-hua, D.; Qiu-ping, Z. Leukemia Stem Cells in Drug Resistance and Metastasis. Chin. Med. J. (Engl.) 2010, 123, 954–960. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Mendelson, A.; Frenette, P.S. Hematopoietic Stem Cell Niche Maintenance during Homeostasis and Regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.-Y.; Jeong, S.-Y.; Lee, H.-R.; Oh, I.-H. Age-Related Differences in the Bone Marrow Stem Cell Niche Generate Specialized Microenvironments for the Distinct Regulation of Normal Hematopoietic and Leukemia Stem Cells. Sci. Rep. 2019, 9, 1007. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lei, W.; Chen, X.; Wang, S.; Qian, W. Oxidative Stress Response Induced by Chemotherapy in Leukemia Treatment (Review). Mol. Clin. Oncol. 2018, 8, 391–399. [Google Scholar] [CrossRef]
- Zhang, C.C.; Sadek, H.A. Hypoxia and Metabolic Properties of Hematopoietic Stem Cells. Antioxid. Redox Signal. 2014, 20, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Lodish, H.F. Cytokines Regulating Hematopoietic Stem Cell Function. Curr. Opin. Hematol. 2008, 15, 307–311. [Google Scholar] [CrossRef]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.-J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive Oxygen Species Regulate Hematopoietic Stem Cell Self-Renewal, Migration and Development, As Well As Their Bone Marrow Microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative Stress and Hypoxia in Normal and Leukemic Stem Cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef]
- Samimi, A.; Kalantari, H.; Lorestani, M.Z.; Shirzad, R.; Saki, N. Oxidative Stress in Normal Hematopoietic Stem Cells and Leukemia. APMIS 2018, 126, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wu, Q.; Zhou, F. Targeting Acute Myeloid Leukemia Stem Cells: Current Therapies in Development and Potential Strategies with New Dimensions. Crit. Rev. Oncol. Hematol. 2020, 152, 102993. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Culp-Hill, R.; Reisz, J.A.; Pei, S.; Gustafson, A.; Khan, N.; DeGregori, J.; Pollyea, D.A.; et al. Cysteine Depletion Targets Leukemia Stem Cells through Inhibition of Electron Transport Complex II. Blood 2019, 134, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef]
- Culp-Hill, R.; D’Alessandro, A.; Pietras, E.M. Extinguishing the Embers: Targeting AML Metabolism. Trends Mol. Med. 2020, S1471491420302628. [Google Scholar] [CrossRef]
- Jordan, C.T. Can We Selectively Target AML Stem Cells? Best Pract. Res. Clin. Haematol. 2019, 32, 101100. [Google Scholar] [CrossRef] [PubMed]
- Bencomo-Alvarez, A.E.; Rubio, A.J.; Gonzalez, M.A.; Eiring, A.M. Energy Metabolism and Drug Response in Myeloid Leukaemic Stem Cells. Br. J. Haematol. 2019, 186, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do Reactive Oxygen Species Play a Role in Myeloid Leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 1–13. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS Signalling: Adaptive Redox Switches through Oxidative/Nitrosative Protein Modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Jayavelu, A.K.; Moloney, J.N.; Böhmer, F.-D.; Cotter, T.G. NOX-Driven ROS Formation in Cell Transformation of FLT3-ITD-Positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-Derived ROS in AML Promotes Proliferation and Is Associated with Defective Oxidative Stress Signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial Metabolism and Cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Basak, N.P.; Banerjee, S. Mitochondrial Dependency in Progression of Acute Myeloid Leukemia. Mitochondrion 2015, 21, 41–48. [Google Scholar] [CrossRef]
- Al Ageeli, E. Alterations of Mitochondria and Related Metabolic Pathways in Leukemia: A Narrative Review. Saudi J. Med. Med. Sci. 2020, 8, 3–11. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Sillar, J.R.; Germon, Z.P.; De Iuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Rinker, L.; Peng, J.; Chilian, W.M. Reactive Oxygen Species: The Good and the Bad. In Reactive Oxygen Species (ROS) in Living Cells; Filip, C., Albu, E., Eds.; InTech: Brisbane, Australia, 2018; ISBN 978-1-78923-134-2. [Google Scholar]
- Mailloux, R.J. Teaching the Fundamentals of Electron Transfer Reactions in Mitochondria and the Production and Detection of Reactive Oxygen Species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial Reactive Oxygen Species and Cancer. Cancer Metab. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Tuffaha, M.S.A.; Guski, H.; Kristiansen, G. Markers and Immunoprofile of Myeloid Neoplasm. In Immunohistochemistry in Tumor Diagnostics; Springer International Publishing: Cham, Switzerland, 2018; pp. 181–184. ISBN 978-3-319-53576-0. [Google Scholar]
- Itonaga, H.; Imanishi, D.; Wong, Y.-F.; Sato, S.; Ando, K.; Sawayama, Y.; Sasaki, D.; Tsuruda, K.; Hasegawa, H.; Imaizumi, Y.; et al. Expression of Myeloperoxidase in Acute Myeloid Leukemia Blasts Mirrors the Distinct DNA Methylation Pattern Involving the Downregulation of DNA Methyltransferase DNMT3B. Leukemia 2014, 28, 1459–1466. [Google Scholar] [CrossRef]
- Nakazato, T.; Sagawa, M.; Yamato, K.; Xian, M.; Yamamoto, T.; Suematsu, M.; Ikeda, Y.; Kizaki, M. Myeloperoxidase Is a Key Regulator of Oxidative Stress–Mediated Apoptosis in Myeloid Leukemic Cells. Clin. Cancer Res. 2007, 13, 5436–5445. [Google Scholar] [CrossRef]
- Malle, E.; Furtmüller, P.G.; Sattler, W.; Obinger, C. Myeloperoxidase: A Target for New Drug Development?: Inhibition of Myeloperoxidase Activity. Br. J. Pharmacol. 2007, 152, 838–854. [Google Scholar] [CrossRef]
- Hosseini, M.; Rezvani, H.R.; Aroua, N.; Bosc, C.; Farge, T.; Saland, E.; Guyonnet-Dupérat, V.; Zaghdoudi, S.; Jarrou, L.; Larrue, C.; et al. Targeting Myeloperoxidase Disrupts Mitochondrial Redox Balance and Overcomes Cytarabine Resistance in Human Acute Myeloid Leukemia. Cancer Res. 2019, 79, 5191–5203. [Google Scholar] [CrossRef]
- Ježek, J.; Cooper, K.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Joshi, A.; Kundu, M. Mitophagy in Hematopoietic Stem Cells: The Case for Exploration. Autophagy 2013, 9, 1737–1749. [Google Scholar] [CrossRef]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Gießl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial Metabolism Contributes to Oxidative Stress and Reveals Therapeutic Targets in Chronic Lymphocytic Leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Gawara, M.M.; Gugalavath, S.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 117727191875539. [Google Scholar] [CrossRef] [PubMed]
- DelRío, L.A.; López-Huertas, E. ROS Generation in Peroxisomes and Its Role in Cell Signaling. Plant. Cell Physiol. 2016, 57, 1364–1376. [Google Scholar] [CrossRef]
- Sandalio, L.M.; Rodríguez-Serrano, M.; Romero-Puertas, M.C.; Del Río, L.A. Role of Peroxisomes as a Source of Reactive Oxygen Species (ROS) Signaling Molecules. In Peroxisomes and their Key Role in Cellular Signaling and Metabolism; Del Río, L.A., Ed.; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2013; Volume 69, pp. 231–255. ISBN 978-94-007-6888-8. [Google Scholar]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian Xanthine Oxidoreductase-Mechanism of Transition from Xanthine Dehydrogenase to Xanthine Oxidase: Mammalian Xanthine Oxidoreductase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Romo-González, M.; Moreno-Paz, S.; García-Hernández, V.; Sánchez-Guijo, F.; Hernández-Hernández, Á. Inhibition of Xanthine Oxidoreductase Enhances the Potential of Tyrosine Kinase Inhibitors against Chronic Myeloid Leukemia. Antioxidants 2020, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.L.; Zhang, W.G.; Wei, Y.C.; Meng, S.; Bai, G.G.; Wang, B.-Y.; Yang, H.-Y.; Tian, W.; Meng, X.; Zhang, H.; et al. Involvement of Oxidative Stress in the Relapse of Acute Myeloid Leukemia. J. Biol. Chem. 2010, 285, 15010–15015. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Hendershot, L.M. Oxidative Folding: Cellular Strategies for Dealing with the Resultant Equimolar Production of Reactive Oxygen Species. Antioxid. Redox Signal. 2009, 11, 2317–2331. [Google Scholar] [CrossRef]
- Dikalov, S.; Dikalova, A.; Bikineyeva, A.; Schmidt, H.; Harrison, D.; Griendling, K. Distinct Roles of Nox1 and Nox4 in Basal and Angiotensin II-Stimulated Superoxide and Hydrogen Peroxide Production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Irwin, M.E.; Rivera-Del Valle, N.; Chandra, J. Redox Control of Leukemia: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 1349–1383. [Google Scholar] [CrossRef] [PubMed]
- Er, T.-K.; Tsai, S.-M.; Wu, S.-H.; Chiang, W.; Lin, H.-C.; Lin, S.-F.; Wu, S.-H.; Tsai, L.-Y.; Liu, T.-Z. Antioxidant Status and Superoxide Anion Radical Generation in Acute Myeloid Leukemia. Clin. Biochem. 2007, 40, 1015–1019. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Battisti, V.; Maders, L.D.K.; Bagatini, M.D.; Santos, K.F.; Spanevello, R.M.; Maldonado, P.A.; Brulé, A.O.; Araújo, M. do C.; Schetinger, M.R.C.; Morsch, V.M. Measurement of Oxidative Stress and Antioxidant Status in Acute Lymphoblastic Leukemia Patients. Clin. Biochem. 2008, 41, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Oltra, A.M.; Carbonell, F.; Tormos, C.; Iradi, A.; Ez, G.T.S. Antioxidant Enzyme Activities and the Production of MDA and 8-Oxo-DG in Chronic Lymphocytic Leukemia. Free Radic. Biol. Med. 2001, 30, 1286–1292. [Google Scholar] [CrossRef]
- Udensi, U.K.; Tchounwou, P.B. Dual Effect of Oxidative Stress on Leukemia Cancer Induction and Treatment. J. Exp. Clin. Cancer Res. 2014, 33, 106. [Google Scholar] [CrossRef]
- Nishiura, T.; Suzuki, K.; Kawaguchi, T.; Nakao, H.; Kawamura, N.; Taniguchi, M.; Kanayama, Y.; Yonezawa, T.; Iizuka, S.; Taniguchi, N. Elevated Serum Manganese Superoxide Dismutase in Acute Leukemias. Cancer Lett. 1992, 62, 211–215. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular Mechanisms and Physiological Consequences of Redox-Dependent Signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide Dismutase as a Target for the Selective Killing of Cancer Cells. Nature 2000, 407, 390–395. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Kan, W.-M. Down-Regulation of Superoxide Dismutase 1 by PMA Is Involved in Cell Fate Determination and Mediated via Protein Kinase D2 in Myeloid Leukemia Cells. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2015, 1853, 2662–2675. [Google Scholar] [CrossRef] [PubMed]
- López-Pedrera, C.; Villalba, J.M.; Siendones, E.; Barbarroja, N.; Gómez-Díaz, C.; Rodríguez-Ariza, A.; Buendía, P.; Torres, A.; Velasco, F. Proteomic Analysis of Acute Myeloid Leukemia: Identification of Potential Early Biomarkers and Therapeutic Targets. Proteomics 2006, 6, S293–S299. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Sethi, G.; Garg, M. The Multifaceted Role of Reactive Oxygen Species in Tumorigenesis. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Lam, C.F.; Yeung, H.T.; Lam, Y.M.; Ng, R.K. Reactive Oxygen Species Activate Differentiation Gene Transcription of Acute Myeloid Leukemia Cells via the JNK/c-JUN Signaling Pathway. Leuk. Res. 2018, 68, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Riccio, P.; Sessa, R.; Nicola, S.; Petruzziello, F.; Trombetti, S.; Menna, G.; Pepe, G.; Maddalena, P.; Izzo, P.; Grosso, M. GATA-1 Isoforms Differently Contribute to the Production and Compartmentation of Reactive Oxygen Species in the Myeloid Leukemia Cell Line K562. J. Cell. Physiol. 2019, 234, 20829–20846. [Google Scholar] [CrossRef]
- Lu, J.; Chew, E.-H.; Holmgren, A. Targeting Thioredoxin Reductase Is a Basis for Cancer Therapy by Arsenic Trioxide. Proc. Natl. Acad. Sci. 2007, 104, 12288–12293. [Google Scholar] [CrossRef]
- Nigro, P.; Dal Piaz, F.; Gallotta, D.; De Tommasi, N.; Belisario, M.A. Inhibition of the Thioredoxin System Is a Basis for the Antileukemic Potential of 13-Hydroxy-15-Oxo-Zoapatlin. Free Radic. Biol. Med. 2008, 45, 875–884. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The Thioredoxin Antioxidant System. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Kuksal, N.; Chalker, J.; Mailloux, R.J. Progress in Understanding the Molecular Oxygen Paradox – Function of Mitochondrial Reactive Oxygen Species in Cell Signaling. Biol. Chem. 2017, 398, 1209–1227. [Google Scholar] [CrossRef]
- Mikhed, Y.; Görlach, A.; Knaus, U.G.; Daiber, A. Redox Regulation of Genome Stability by Effects on Gene Expression, Epigenetic Pathways and DNA Damage/Repair. Redox Biol. 2015, 5, 275–289. [Google Scholar] [CrossRef]
- García-Guede, Á.; Vera, O.; Ibáñez-de-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive Oxygen Species in Cancer Stem Cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Rathore, M.G.; Allende-Vega, N.; Vo, D.-N.; Belkhala, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; Lecellier, C.-H.; et al. Human Leukemic Cells Performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen Species (ROS) Production. EBioMedicine 2016, 3, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Montano, G.; Vidovic, K.; Palladino, C.; Cesaro, E.; Sodaro, G.; Quintarelli, C.; De Angelis, B.; Errichiello, S.; Pane, F.; Izzo, P.; et al. WT1-Mediated Repression of the Proapoptotic Transcription Factor ZNF224 Is Triggered by the BCR-ABL Oncogene. Oncotarget 2015, 6, 28223–28237. [Google Scholar] [CrossRef]
- Kreuz, S.; Fischle, W. Oxidative Stress Signaling to Chromatin in Health and Disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef]
- Tothova, Z.; Gilliland, D.G. FoxO Transcription Factors and Stem Cell Homeostasis: Insights from the Hematopoietic System. Cell Stem Cell 2007, 1, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gan, B.; Liu, D.; Paik, J. FoxO Family Members in Cancer. Cancer Biol. Ther. 2011, 12, 253–259. [Google Scholar] [CrossRef]
- Menon, V.; Ghaffari, S. Transcription Factors FOXO in the Regulation of Homeostatic Hematopoiesis. Curr. Opin. Hematol. 2018, 25, 290–298. [Google Scholar] [CrossRef]
- Liang, R.; Ghaffari, S. Mitochondria and FOXO3 in Stem Cell Homeostasis, a Window into Hematopoietic Stem Cell Fate Determination. J. Bioenerg. Biomembr. 2017, 49, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A Directs a Protective Autophagy Program in Haematopoietic Stem Cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Liu, H.; Luo, X.-J.; Zhao, Z.; Zou, Z.-Y.; Li, J.; Lin, X.-J.; Liang, Y. The Roles of Reactive Oxygen Species (ROS) and Autophagy in the Survival and Death of Leukemia Cells. Crit. Rev. Oncol. Hematol. 2017, 112, 21–30. [Google Scholar] [CrossRef]
- Gurnari, C.; Falconi, G.; De Bellis, E.; Voso, M.T.; Fabiani, E. The Role of Forkhead Box Proteins in Acute Myeloid Leukemia. Cancers 2019, 11, 865. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Copple, I.M.; Goldring, C.E.; Kitteringham, N.R.; Park, B.K. The Keap1-Nrf2 Cellular Defense Pathway: Mechanisms of Regulation and Role in Protection Against Drug-Induced Toxicity. In Adverse Drug Reactions; Uetrecht, J., Ed.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2010; Volume 196, pp. 233–266. ISBN 978-3-642-00662-3. [Google Scholar]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global Mapping of Binding Sites for Nrf2 Identifies Novel Targets in Cell Survival Response through ChIP-Seq Profiling and Network Analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 System in Development, Oxidative Stress Response and Diseases: An Evolutionarily Conserved Mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Kwong, M. Impaired Expression of Glutathione Synthetic Enzyme Genes in Mice with Targeted Deletion of the Nrf2 Basic-Leucine Zipper Protein. Biochim. Biophys. Acta 2000, 1517, 19–26. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in Cancers: A Double-edged Sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E. The Keap1-Nrf2 Pathway: Mechanisms of Activation and Dysregulation in Cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, A.; MacEwan, D.J.; Bowles, K.M.; Rushworth, S.A. Oxidative Stress Responses and NRF2 in Human Leukaemia. Oxid. Med. Cell. Longev. 2015, 2015, 454659. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The High Nrf2 Expression in Human Acute Myeloid Leukemia Is Driven by NF-ΚB and Underlies Its Chemo-Resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.J.; Baldwin, A.S. NF-ΚB Suppresses ROS Levels in BCR–ABL+ Cells to Prevent Activation of JNK and Cell Death. Oncogene 2011, 30, 4557–4566. [Google Scholar] [CrossRef]
- Franklin, R.; McCubrey, J. Kinases: Positive and Negative Regulators of Apoptosis. Leukemia 2000, 14, 2019–2034. [Google Scholar] [CrossRef]
- Deynoux, M.; Sunter, N.; Hérault, O.; Mazurier, F. Hypoxia and Hypoxia-Inducible Factors in Leukemias. Front. Oncol. 2016, 6, 41. [Google Scholar] [CrossRef]
- Wielockx, B.; Grinenko, T.; Mirtschink, P.; Chavakis, T. Hypoxia Pathway Proteins in Normal and Malignant Hematopoiesis. Cells 2019, 8, 155. [Google Scholar] [CrossRef]
- Wierenga, A.T.J.; Cunningham, A.; Erdem, A.; Lopera, N.V.; Brouwers-Vos, A.Z.; Pruis, M.; Mulder, A.B.; Günther, U.L.; Martens, J.H.A.; Vellenga, E.; et al. HIF1/2-Exerted Control over Glycolytic Gene Expression Is Not Functionally Relevant for Glycolysis in Human Leukemic Stem/Progenitor Cells. Cancer Metab. 2019, 7, 11. [Google Scholar] [CrossRef]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and Cancer Stem Cells: Molecular Mechanisms and Therapeutic Applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef]
- Qian, Q.; Chen, W.; Cao, Y.; Cao, Q.; Cui, Y.; Li, Y.; Wu, J. Targeting Reactive Oxygen Species in Cancer via Chinese Herbal Medicine. Oxid. Med. Cell. Longev. 2019, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Kim, M.; Park, H.; Jeong, M.I.; Jung, W.; Kim, B. Natural Products and Acute Myeloid Leukemia: A Review Highlighting Mechanisms of Action. Nutrients 2019, 11, 1010. [Google Scholar] [CrossRef]
- Zhou, F.; Shen, Q.; Claret, F.X. Novel Roles of Reactive Oxygen Species in the Pathogenesis of Acute Myeloid Leukemia. J. Leukoc. Biol. 2013, 94, 423–429. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing Leukemia Stem Cells to NF-ΚB Inhibitor Treatment in Vivo by Inactivation of Both TNF and IL-1 Signaling. Oncotarget 2017, 8, 8420–8435. [Google Scholar] [CrossRef] [PubMed]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of Mitochondrial Electron Transport Chain Function Potentiates the Pro-Apoptotic Effects of MAPK Inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef]
- Donovan, M.; Cotter, T.G. Control of Mitochondrial Integrity by Bcl-2 Family Members and Caspase-Independent Cell Death. Biochim. Biophys. Acta 2004, 1644, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Patients with Acute Myeloid Leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Dong, J.; Liu, B.; Zhu, R. Targeting ROS for Cancer Therapy. Chemotherapy 2016, 5. [Google Scholar] [CrossRef]
- Sieber, M.A.; Hegel, J.K.E. Azelaic Acid: Properties and Mode of Action. Skin Pharmacol. Physiol. 2014, 27, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Luo, Z.; Jin, Y.; Chen, Y.; Yang, T.; Yang, Q.; Wu, B.; Shang, Y.; Liu, X.; Wei, Y.; et al. Azelaic Acid Exerts Antileukemia Effects against Acute Myeloid Leukemia by Regulating the Prdxs/ROS Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Palumbo, I.; Grosso, M.; Slater, N.; Miles, C.G. WT1 Regulates Murine Hematopoiesis via Maintenance of VEGF Isoform Ratio. Blood 2013, 122, 188–192. [Google Scholar] [CrossRef]
- Zhe, N.; Chen, S.; Zhou, Z.; Liu, P.; Lin, X.; Yu, M.; Cheng, B.; Zhang, Y.; Wang, J. HIF-1α Inhibition by 2-Methoxyestradiol Induces Cell Death via Activation of the Mitochondrial Apoptotic Pathway in Acute Myeloid Leukemia. Cancer Biol. Ther. 2016, 17, 625–634. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, J.; Zeng, H.; Zhang, Y.; Zhang, Y.; Zhou, X.; Zhou, H. Antiproliferative Effects of L-asparaginase in Acute Myeloid Leukemia. Exp. Ther. Med. 2020. [Google Scholar] [CrossRef]
- Tibodeau, J.D.; Benson, L.M.; Isham, C.R.; Owen, W.G.; Bible, K.C. The Anticancer Agent Chaetocin Is a Competitive Substrate and Inhibitor of Thioredoxin Reductase. Antioxid. Redox Signal. 2009, 11, 1097–1106. [Google Scholar] [CrossRef]
- Lai, Y.-S.; Chen, J.-Y.; Tsai, H.-J.; Chen, T.-Y.; Hung, W.-C. The SUV39H1 Inhibitor Chaetocin Induces Differentiation and Shows Synergistic Cytotoxicity with Other Epigenetic Drugs in Acute Myeloid Leukemia Cells. Blood Cancer J. 2015, 5, e313. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trombetti, S.; Cesaro, E.; Catapano, R.; Sessa, R.; Lo Bianco, A.; Izzo, P.; Grosso, M. Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 2470. https://doi.org/10.3390/ijms22052470
Trombetti S, Cesaro E, Catapano R, Sessa R, Lo Bianco A, Izzo P, Grosso M. Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia. International Journal of Molecular Sciences. 2021; 22(5):2470. https://doi.org/10.3390/ijms22052470
Chicago/Turabian StyleTrombetti, Silvia, Elena Cesaro, Rosa Catapano, Raffaele Sessa, Alessandra Lo Bianco, Paola Izzo, and Michela Grosso. 2021. "Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia" International Journal of Molecular Sciences 22, no. 5: 2470. https://doi.org/10.3390/ijms22052470
APA StyleTrombetti, S., Cesaro, E., Catapano, R., Sessa, R., Lo Bianco, A., Izzo, P., & Grosso, M. (2021). Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia. International Journal of Molecular Sciences, 22(5), 2470. https://doi.org/10.3390/ijms22052470