
Targeting AKT/mTOR and Bcl-2 for Autophagic and Apoptosis Cell Death in Lung Cancer: Novel Activity of a Polyphenol Compound

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Test Compound
2.2. Cell Culture
2.3. Chemicals
2.4. Cytotoxicity Assays
2.5. Cell Death Assay
2.6. Flow Cytometry for Apoptosis
2.7. Western Blot Analysis
2.8. Transmission Electron Microscopy
2.9. Monodansylcadaverine Staining
2.10. Immunofluorescence
2.11. Mass Spectrometry-Based Proteomics
2.12. Small Interfering RNA (siRNA) Transfection
2.13. Patient-Derived Primary Lung Cancer Cell Line from Malignant Pleural Effusion
2.14. Statistical Analysis
3. Results
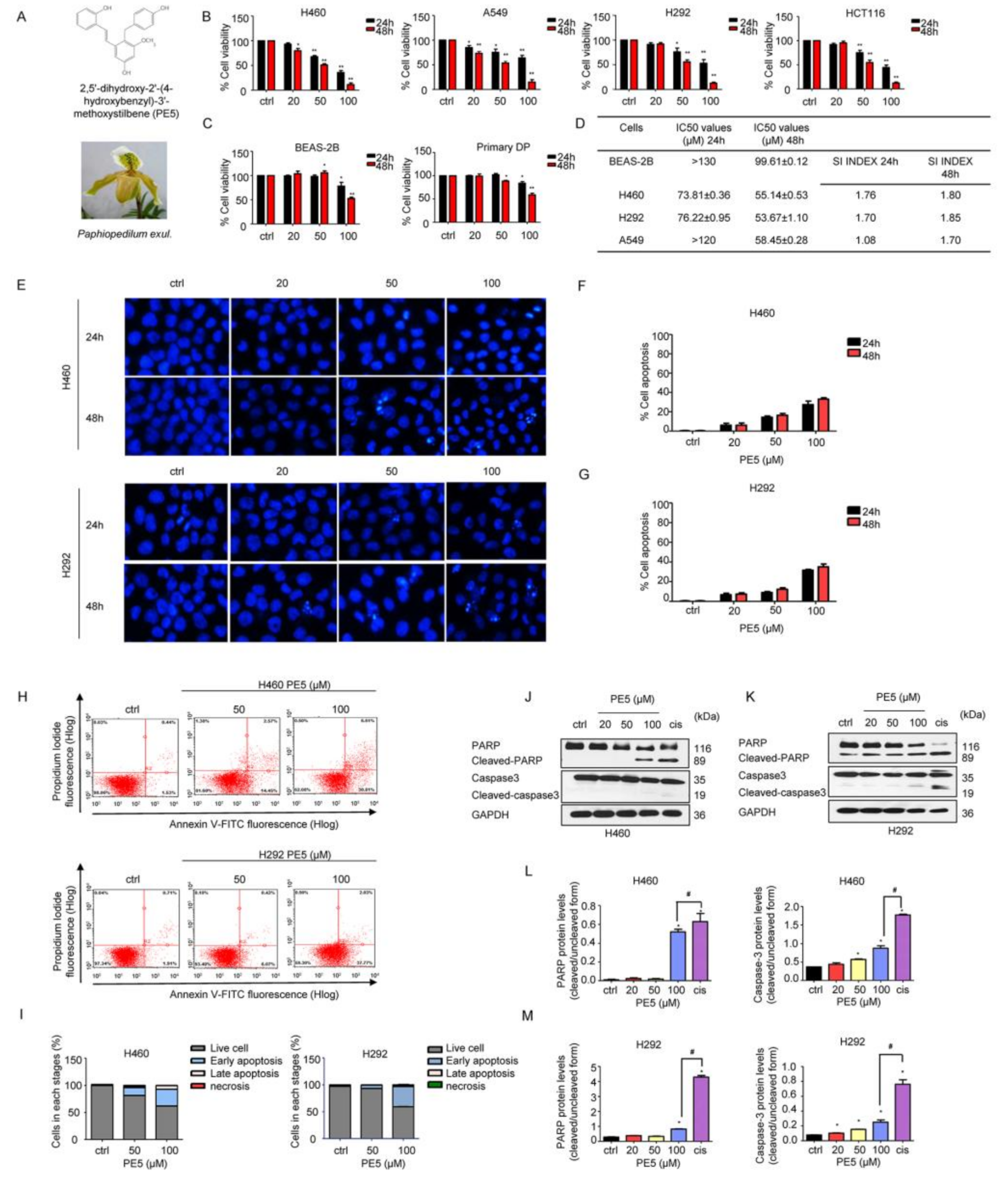
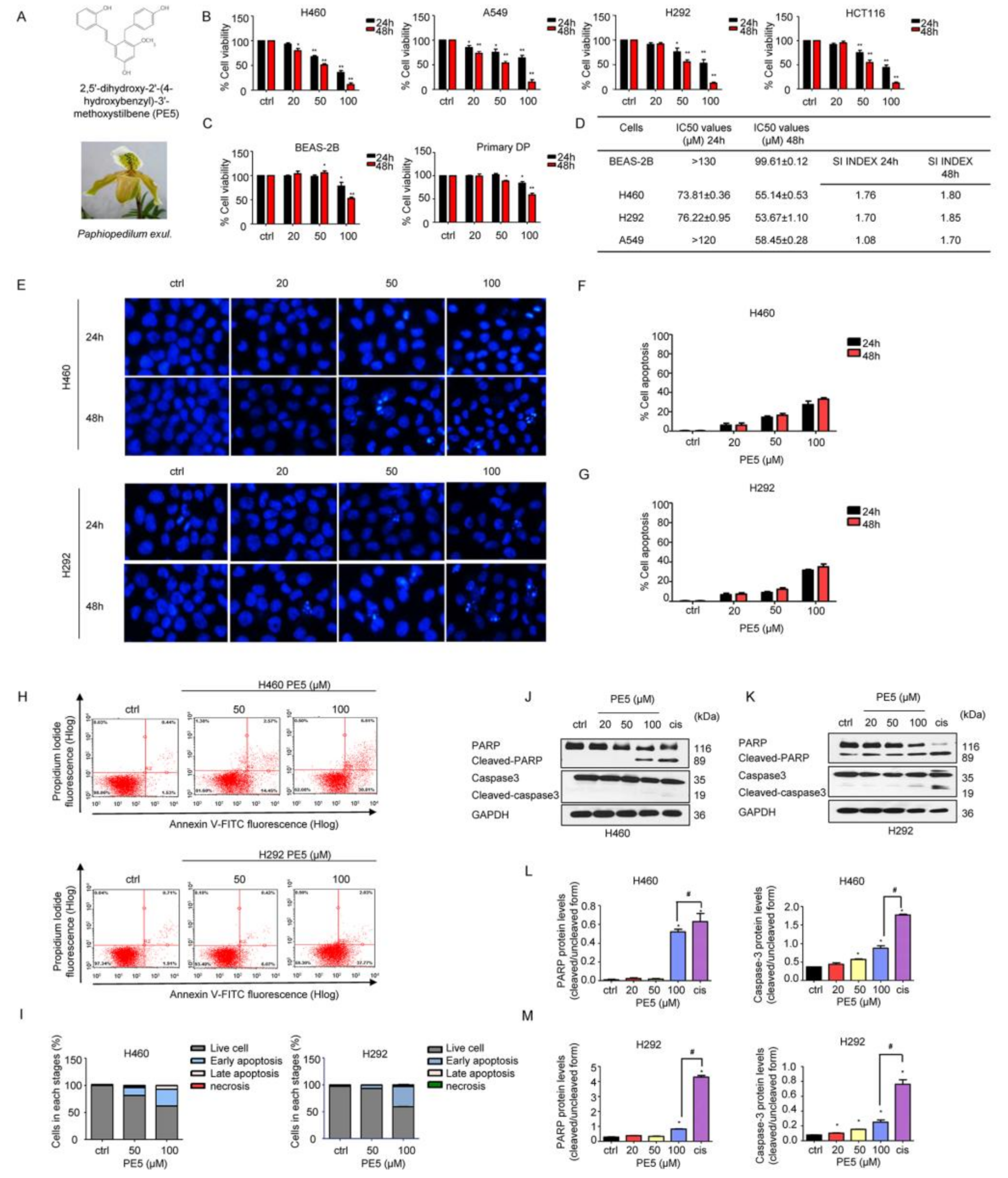
3.1. Anti-Cancer Activities of PE5 on Lung Cancer Cells
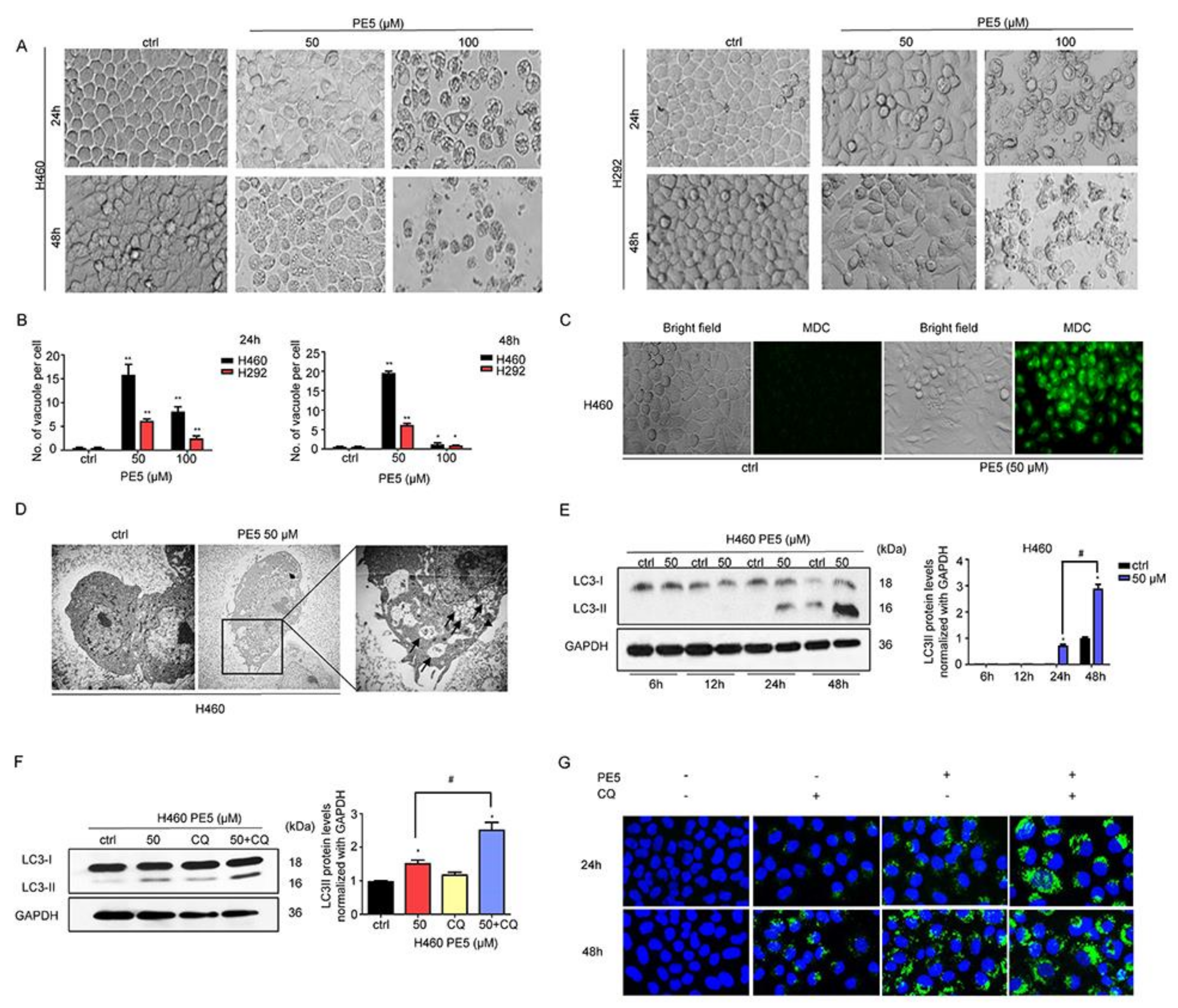
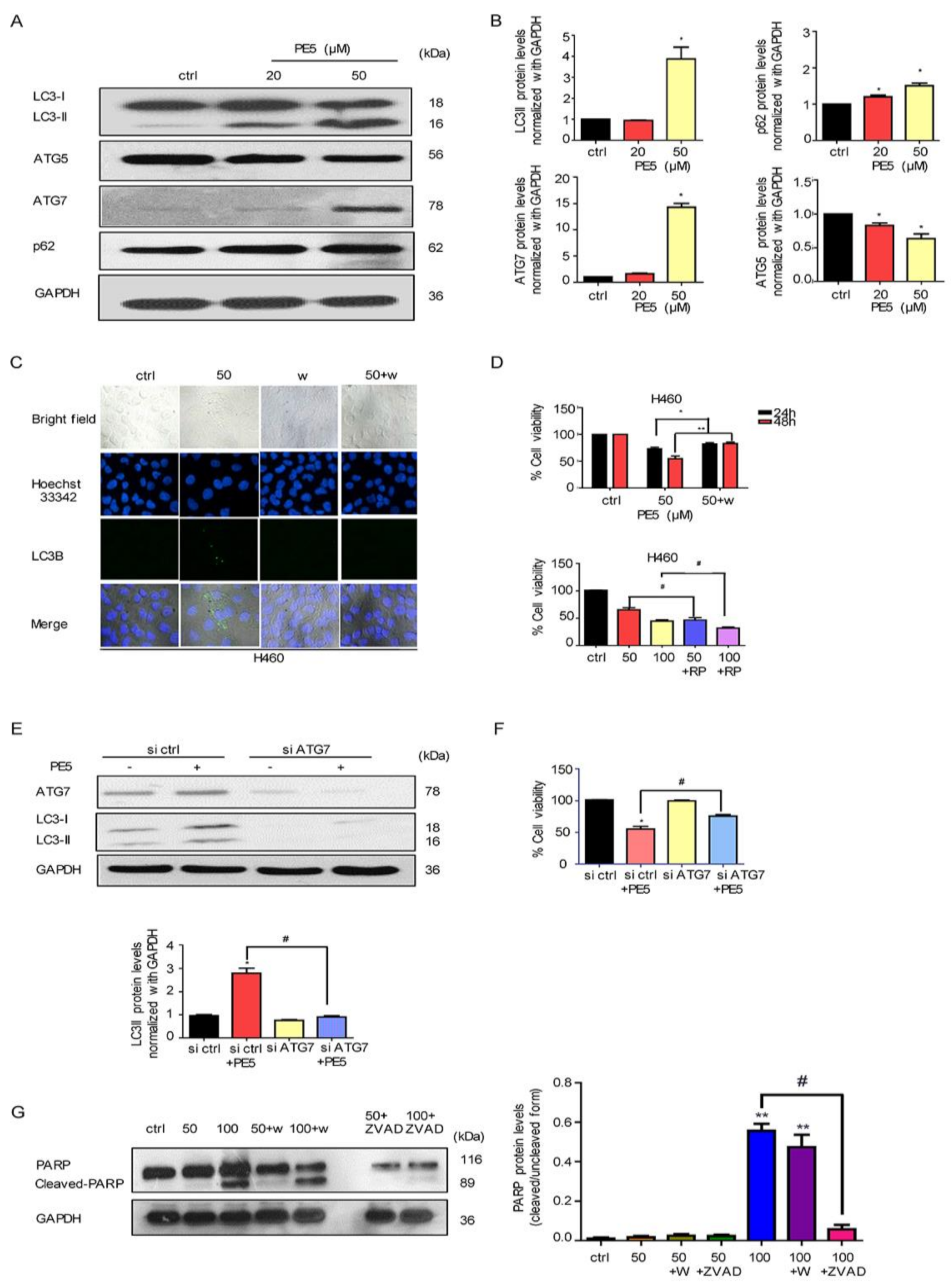
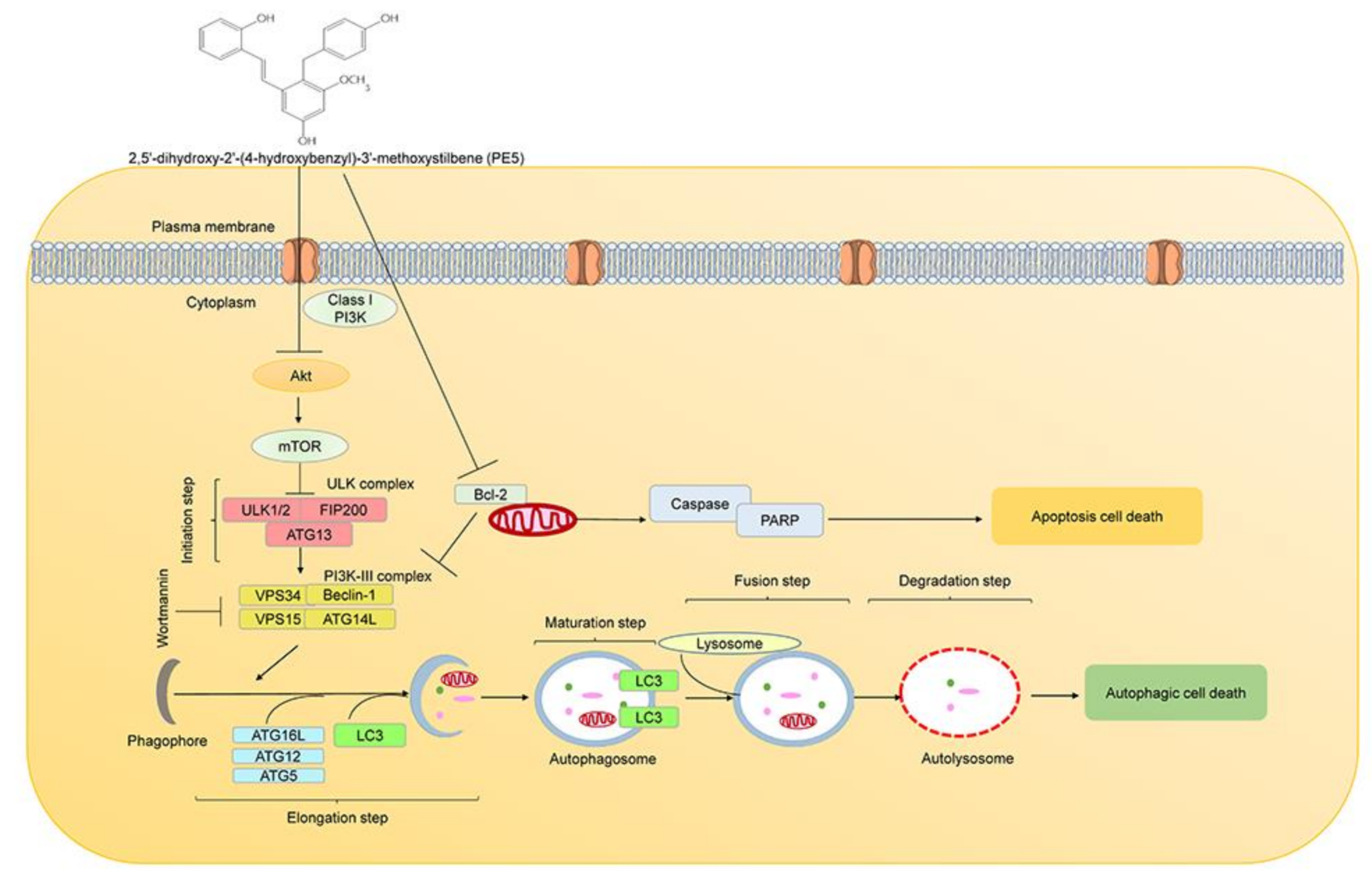
3.2. PE5-Induced Autophagy in Lung Cancer Cells
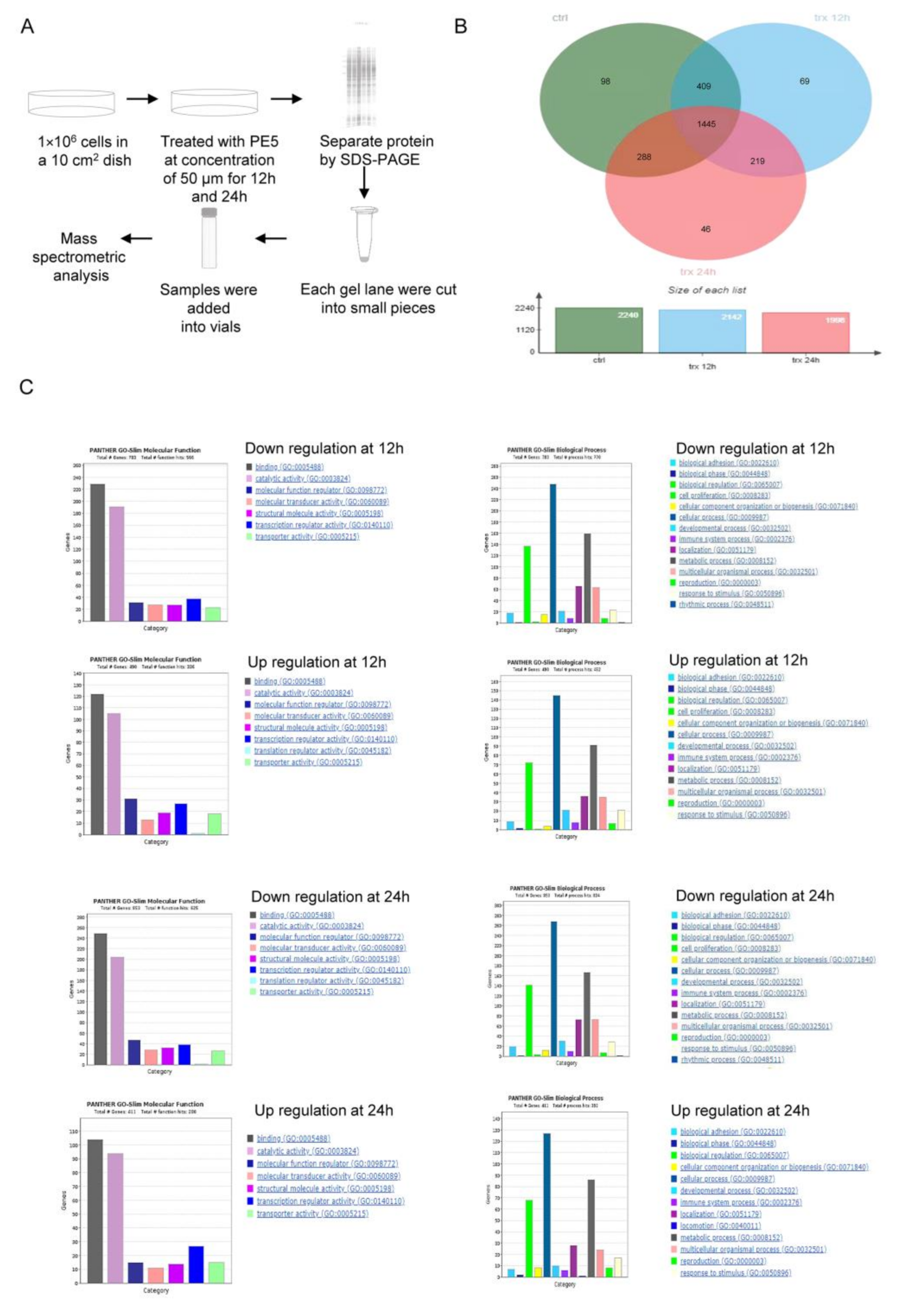
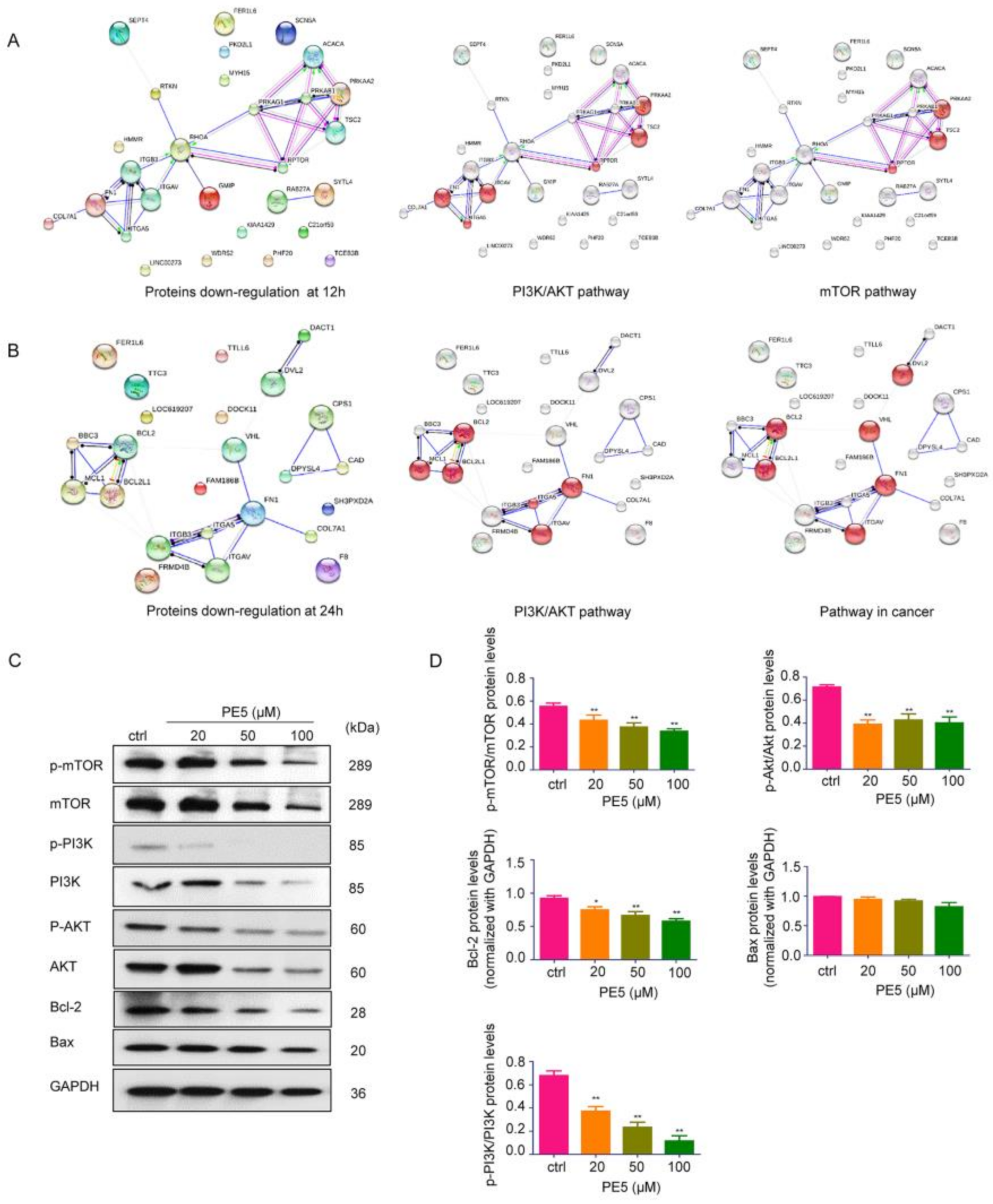
3.3. Molecular Functions and Biological Processes of the Proteins in PE5-Treated Cells
3.4. Mechanisms of Action of PE5 Analyzed by the Protein–Protein Interaction Networks and Signaling Pathways
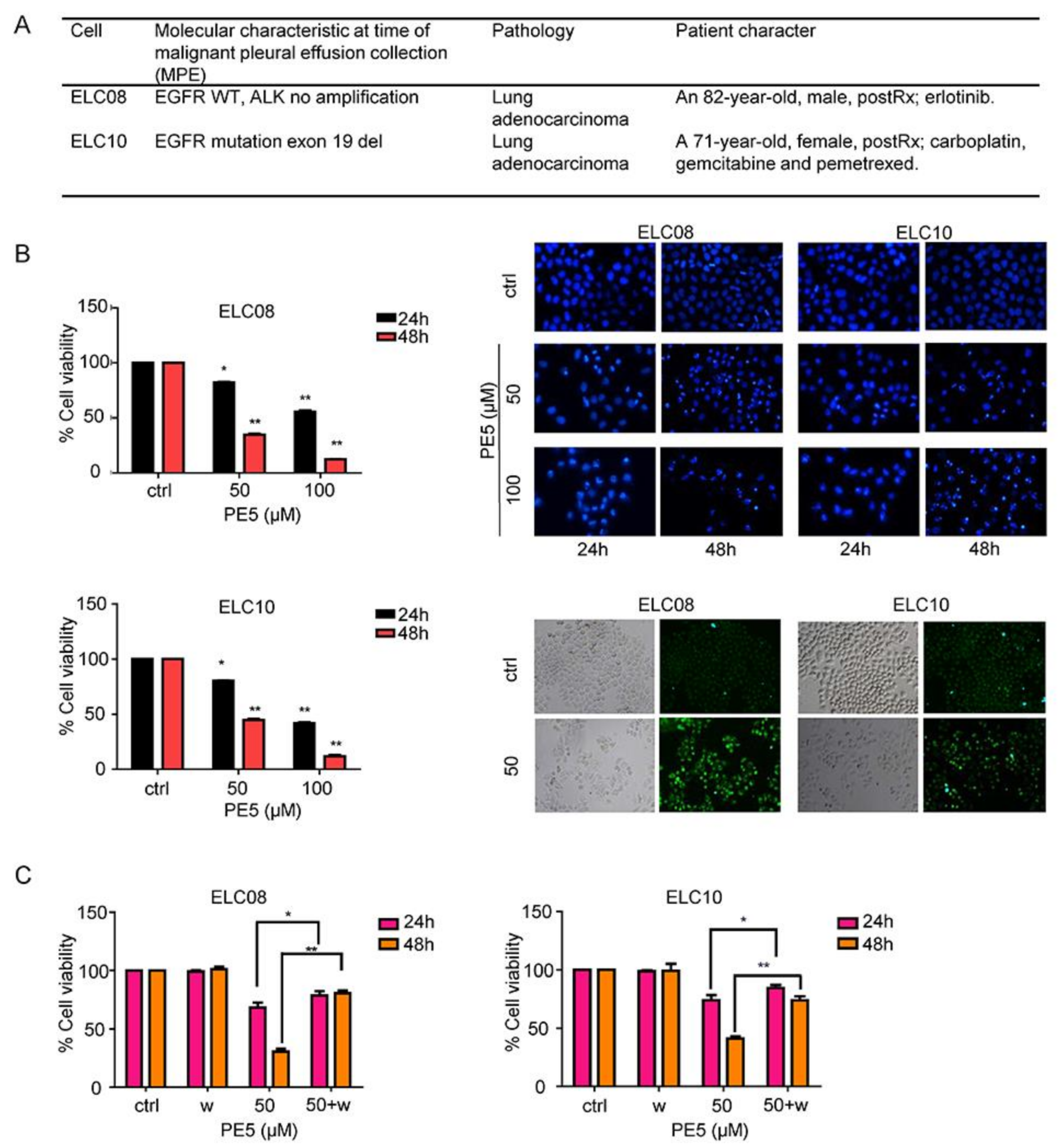
3.5. Effect of PE5 on Patient-Derived Primary Lung Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Groot, P.M.; Wu, C.C.; Carter, B.W.; Munden, R.F. The epidemiology of lung cancer. Transl. Lung Cancer Res. 2018, 7, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Kim, E.S. Chemotherapy Resistance in Lung Cancer. Adv. Exp. Med. Biol. 2016, 893, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Plati, J.; Bucur, O.; Khosravi-Far, R. Dysregulation of apoptotic signaling in cancer: Molecular mechanisms and therapeutic opportunities. J. Cell. Biochem. 2008, 104, 1124–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Deiry, W.S. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003, 22, 7486–7495. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Kondo, S. Autophagy and cancer therapy. Autophagy 2006, 2, 85–90. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, B.Y.K.; Chan, W.K.; Xu, S.W.; Wang, J.R.; Bai, L.P.; Liu, L.; Wong, V.K.W. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci. Rep. 2014, 4, 5510. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhou, Y.; Cheng, X.; Fan, Y.; He, S.; Li, S.; Ye, H.; Xie, C.; Wu, W.; Li, C.; et al. Isogambogenic acid induces apoptosis-independent autophagic cell death in human non-small-cell lung carcinoma cells. Sci. Rep. 2015, 5, 7697. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.D.; Kang, K.A.; Kim, H.S.; Kim, D.H.; Choi, Y.H.; Lee, S.J.; Kim, H.S.; Hyun, J.W. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013, 4, e750. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.J.; Zhang, Y.M.; Zhang, L.; Huang, B.; Tao, F.F.; Chen, W.; Guo, Z.J.; Xu, Q.; Sun, Y. Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy 2013, 9, 996–1008. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Salunke-Gawalib, S.; Konkimallaa, V.B. Induction of Autophagic Cell Death in Apoptosis-resistant Pancreatic Cancer Cells using Benzo[alpha]phenoxazines Derivatives, 10-methyl-benzo[alpha]phenoxazine-5-one and benzo[alpha]phenoxazine-5-one. Anti-Cancer Agents Med. Chem. 2017, 17, 115–125. [Google Scholar]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.C.; He, S.M.; He, Z.X.; Li, M.; Yang, Y.; Pang, J.X.; Zhang, X.; Chow, K.; Zhou, Q.; Duan, W.; et al. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 2014, 344, 239–259. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapamycin for longevity: Opinion article. Aging 2019, 11, 8048–8067. [Google Scholar] [CrossRef]
- Akar, U.; Chaves-Reyez, A.; Barria, M.; Tari, A.; Sanguino, A.; Kondo, Y.; Kondo, S.; Arun, B.; Lopez-Berestein, G.; Ozpolat, B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 2008, 4, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [Green Version]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.-P.; Parys, J.B.; Bultynck, G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef] [Green Version]
- Minh, T.N.; Khang, D.T.; Tuyen, P.T.; Minh, L.T.; Anh, L.H.; Quan, N.V.; Ha, P.T.T.; Quan, N.T.; Toan, N.P.; Elzaawely, A.A.; et al. Phenolic Compounds and Antioxidant Activity of Phalaenopsis Orchid Hybrids. Antioxidants 2016, 5, 31. [Google Scholar] [CrossRef]
- Auberon, F.; Olatunji, J.O.; Krisa, S.; Antheaume, C.; Herbette, G.; Bonté, F.; Mérillon, J.-M.; Lobstein, A. Two New Stilbenoids from the Aerial Parts of Arundina graminifolia (Orchidaceae). Molecules 2016, 21, 1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garo, E.; Hu, J.F.; Goering, M.; Hough, G.; O’Neil-Johnson, M.; Eldridge, G. Stilbenes from the Orchid Phragmipedium sp. J. Nat. Prod. 2007, 70, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Okuda, H. Resveratrol isolated from Polygonum cuspidatum root prevents tumor growth and metastasis to lung and tumor-induced neovascularization in Lewis lung carcinoma-bearing mice. J. Nutr. 2001, 131, 1844–1849. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhou, J.; Jiang, Y. Resveratrol in lung cancer- a systematic review. JBUON 2016, 21, 950–953. [Google Scholar] [PubMed]
- De Filippis, B.; Ammazzalorso, A.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R. Anticancer Activity of Stilbene-Based Derivatives. ChemMedChem 2017, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.S.; Lee, H.J.; Kim, S.H.; Lee, E.O. Piceatannol suppresses breast cancer cell invasion through the inhibition of MMP-9: Involvement of PI3K/AKT and NF-kappaB pathways. J. Agric. Food Chem. 2012, 60, 4083–4089. [Google Scholar] [CrossRef]
- Shen, C.-H.; Shee, J.-J.; Wu, J.-Y.; Lin, Y.-W.; Wu, J.-D.; Liu, Y.-W. Combretastatin A-4 inhibits cell growth and metastasis in bladder cancer cells and retards tumour growth in a murine orthotopic bladder tumour model. Br. J. Pharmacol. 2010, 160, 2008–2027. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Gulcicek, E.E.; Colangelo, C.M.; McMurray, W.; Stone, K.; Williams, K.; Wu, T.; Zhao, H.; Spratt, H.; Kurosky, A.; Wu, B. Proteomics and the analysis of proteomic data: An overview of current protein-profiling technologies. Curr. Protoc. Bioinform. 2005, 10, 13.1.1–13.1.31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosaert, J.; Quoix, E. Platinum drugs in the treatment of non-small-cell lung cancer. Br. J. Cancer 2002, 87, 825–833. [Google Scholar] [CrossRef] [Green Version]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Hernández, M.; Arias, A.; Martínez-García, D.; Pérez-Tomás, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death—Where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131, jcs215152. [Google Scholar] [CrossRef] [Green Version]
- Law, B.Y.K.; Michelangeli, F.; Qu, Y.Q.; Xu, S.-W.; Han, Y.; Mok, S.W.F.; Dias, I.R.D.S.R.; Javed, M.-U.-H.; Chan, W.-K.; Xue, W.-W.; et al. Neferine induces autophagy-dependent cell death in apoptosis-resistant cancers via ryanodine receptor and Ca2+-dependent mechanism. Sci. Rep. 2019, 9, 20034. [Google Scholar] [CrossRef] [PubMed]
- Law, B.Y.K.; Mok, S.W.F.; Chen, J.; Michelangeli, F.; Jiang, Z.-H.; Han, Y.; Qu, Y.Q.; Qiu, A.C.L.; Xu, S.-W.; Xue, W.-W.; et al. N-Desmethyldauricine Induces Autophagic Cell Death in Apoptosis-Defective Cells via Ca(2+) Mobilization. Front. Pharmacol. 2017, 8, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Chávez, J.; Fonseca-Sánchez, M.A.; Arechaga-Ocampo, E.; Flores-Pérez, A.; Palacios-Rodríguez, Y.; Domínguez-Gómez, G.; Marchat, L.A.; Fuentes-Mera, L.; Mendoza-Hernández, G.; Gariglio, P.; et al. Proteomic profiling reveals that resveratrol inhibits HSP27 expression and sensitizes breast cancer cells to doxorubicin therapy. PLoS ONE 2013, 8, e64378. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.-H.; Liu, L.-Z. Role of mTOR in anticancer drug resistance: Perspectives for improved drug treatment. Drug Resist. Updat. 2008, 11, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. S2), 1509–1518. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wold, M.S.; Lim, J.; Lachance, V.; Deng, Z.; Yue, Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol. Neurodegener. 2016, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Res. 2008, 36, D684–D688. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.T.; Xu, L. Bcl-2:Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar]
- Yue, Z.; Horton, A.; Bravin, M.; DeJager, P.L.; Selimi, F.; Heintz, N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: Implications for neurodegeneration in lurcher mice. Neuron 2002, 35, 921–933. [Google Scholar] [CrossRef] [Green Version]
- Tai, W.T.; Shiau, C.W.; Chen, H.L.; Liu, C.Y.; Lin, C.S.; Cheng, A.L.; Chen, P.J.; Chen, K.F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef]
- Rahman, M.A.; Bishayee, K.; Sadra, A.; Huh, S.-O. Oxyresveratrol activates parallel apoptotic and autophagic cell death pathways in neuroblastoma cells. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 23–36. [Google Scholar] [CrossRef]
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 2018, 5, 87. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.-G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Shi, A.; Wang, N.; Li, M.; He, X.; Yin, C.; Tu, Q.; Shen, X.; Tao, Y.; Wang, Q.; et al. Polyphenolic Proanthocyanidin-B2 suppresses proliferation of liver cancer cells and hepatocellular carcinogenesis through directly binding and inhibiting AKT activity. Redox Biol. 2020, 37, 101701. [Google Scholar] [CrossRef] [PubMed]
- Abbas, G.M.; Abdel Bar, F.M.; Baraka, H.N.; Gohar, A.A.; Lahloub, M.F. A new antioxidant stilbene and other constituents from the stem bark of Morus nigra L. Nat. Prod. Res. 2014, 28, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Ovesná, Z.; Kozics, K.; Bader, Y.; Saiko, P.; Handler, N.; Erker, T.; Szekeres, T. Antioxidant activity of resveratrol, piceatannol and 3,3’,4,4’,5,5’-hexahydroxy-trans-stilbene in three leukemia cell lines. Oncol. Rep. 2006, 16, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinisalo, M.; Kårlund, A.; Koskela, A.; Kaarniranta, K.; Karjalainen, R.O. Polyphenol Stilbenes: Molecular Mechanisms of Defence against Oxidative Stress and Aging-Related Diseases. Oxidative Med. Cell. Longev. 2015, 2015, 340520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Li, J.; Yang, Y.; Zhao, X.; Liu, Y.; Jiang, Y.; Zhou, L.; Feng, Y.; Yu, Y.; Cheng, Y. Resveratrol modulates the apoptosis and autophagic death of human lung adenocarcinoma A549 cells via a p53‑dependent pathway: Integrated bioinformatics analysis and experimental validation. Int. J. Oncol. 2020, 57, 925–938. [Google Scholar] [CrossRef]
- Zhang, J.; Chiu, J.; Zhang, H.; Qi, T.; Tang, Q.; Ma, K.; Lu, H.; Li, G. Autophagic cell death induced by resveratrol depends on the Ca2+/AMPK/mTOR pathway in A549 cells. Biochem. Pharmacol. 2013, 86, 317–328. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Sukumaran, P.; Singh, B.B. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol. Carcinog. 2016, 55, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Musial, C.; Siedlecka-Kroplewska, K.; Kmiec, Z.; Gorska-Ponikowska, M. Modulation of Autophagy in Cancer Cells by Dietary Polyphenols. Antioxidants 2021, 10, 123. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tungsukruthai, S.; Reamtong, O.; Roytrakul, S.; Sukrong, S.; Vinayanwattikun, C.; Chanvorachote, P. Targeting AKT/mTOR and Bcl-2 for Autophagic and Apoptosis Cell Death in Lung Cancer: Novel Activity of a Polyphenol Compound. Antioxidants 2021, 10, 534. https://doi.org/10.3390/antiox10040534
Tungsukruthai S, Reamtong O, Roytrakul S, Sukrong S, Vinayanwattikun C, Chanvorachote P. Targeting AKT/mTOR and Bcl-2 for Autophagic and Apoptosis Cell Death in Lung Cancer: Novel Activity of a Polyphenol Compound. Antioxidants. 2021; 10(4):534. https://doi.org/10.3390/antiox10040534
Chicago/Turabian StyleTungsukruthai, Sucharat, Onrapak Reamtong, Sittiruk Roytrakul, Suchada Sukrong, Chanida Vinayanwattikun, and Pithi Chanvorachote. 2021. "Targeting AKT/mTOR and Bcl-2 for Autophagic and Apoptosis Cell Death in Lung Cancer: Novel Activity of a Polyphenol Compound" Antioxidants 10, no. 4: 534. https://doi.org/10.3390/antiox10040534
APA StyleTungsukruthai, S., Reamtong, O., Roytrakul, S., Sukrong, S., Vinayanwattikun, C., & Chanvorachote, P. (2021). Targeting AKT/mTOR and Bcl-2 for Autophagic and Apoptosis Cell Death in Lung Cancer: Novel Activity of a Polyphenol Compound. Antioxidants, 10(4), 534. https://doi.org/10.3390/antiox10040534