
Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity


{kind=link}
{kind=link}
Abstract
:1. Introduction
Primer on Skeletal Muscle Wasting
2. Signaling Links between Mitochondrial Dysfunction and Skeletal Muscle Wasting
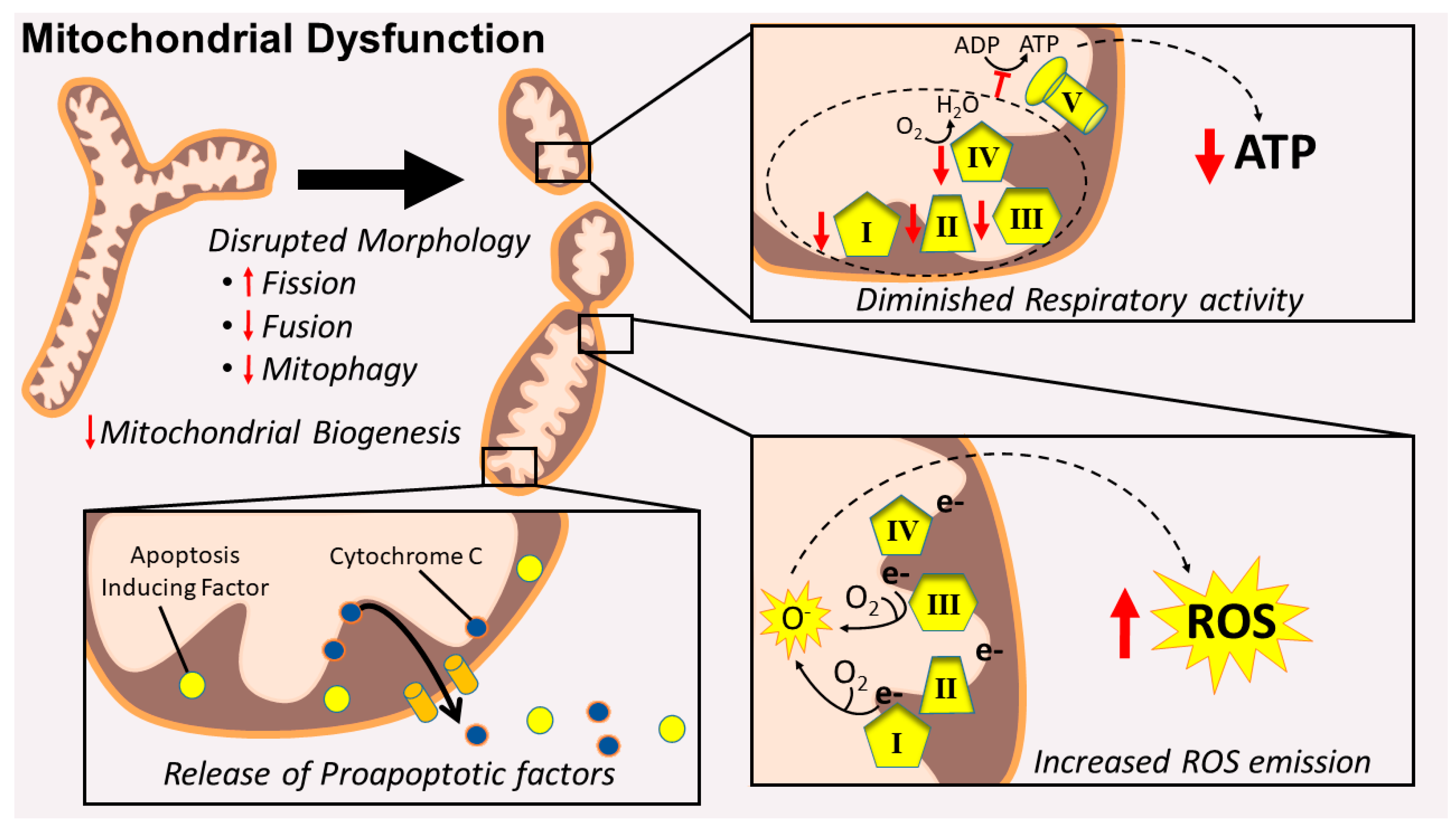
2.1. Mitochondrial Signaling Leading to Skeletal Muscle Wasting: Premise
2.2. Mitochondrial Damage Results in the Release of Proapoptotic Factors
2.3. Mitochondrial Dysfunction Results in Energy Stress
3. Mitochondrial Dysfunction and Skeletal Muscle Atrophy
Mitochondrial Dysfunction Resulting in Increased Mitochondrial Ros Emission Promotes Inactivity-Induced Muscle Atrophy
4. Mitochondrial Dysfunction and Sarcopenia
5. Role of Mitochondrial Dysfunction in Chemotherapy-Induced Muscle Wasting
6. Mitochondria and Cancer Cachexia
7. Evidence Linking Mitochondrial Dysfunction with Sepsis-Induced Muscle Wasting
8. Summary and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Febbraio, M.A.; Hiscock, N.; Sacchetti, M.; Fischer, C.P.; Pedersen, B.K. Interleukin-6 is a novel factor mediating glucose homeostasis during skeletal muscle contraction. Diabetes 2004, 53, 1643–1648. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Dostou, J.M.; Welle, S.L.; Gerich, J.E. Role of human liver, kidney, and skeletal muscle in postprandial glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E419–E427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikanthan, P.; Karlamangla, A.S. Muscle mass index as a predictor of longevity in older adults. Am. J. Med. 2014, 127, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Weijs, P.J.; Looijaard, W.G.; Dekker, I.M.; Stapel, S.N.; Girbes, A.R.; Oudemans-van Straaten, H.M.; Beishuizen, A. Low skeletal muscle area is a risk factor for mortality in mechanically ventilated critically ill patients. Crit. Care 2014, 18, R12. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Edward, F. Adolph Distinguished Lecture. Skeletal muscle atrophy: Multiple pathways leading to a common outcome. J. Appl. Physiol. 2020, 129, 272–282. [Google Scholar] [CrossRef]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef] [PubMed]
- Dupont-Versteegden, E.E.; McCarthy, J.J. Translational control of muscle mass. J. Appl. Physiol. 2019, 127, 579–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [Green Version]
- Tian, Q.; Stepaniants, S.B.; Mao, M.; Weng, L.; Feetham, M.C.; Doyle, M.J.; Yi, E.C.; Dai, H.; Thorsson, V.; Eng, J.; et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol. Cell. Proteomics 2004, 3, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, R.; Jensen, T.E.; Goodman, C.A.; Hornberger, T.A. Resistance Exercise-Induced Hypertrophy: A Potential Role for Rapamycin-Insensitive mTOR. Exerc. Sport Sci. Rev. 2019, 47, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.B.; Smuder, A.J.; Nelson, W.B.; Wiggs, M.P.; Shimkus, K.L.; Fluckey, J.D.; Szeto, H.H.; Powers, S.K. Partial Support Ventilation and Mitochondrial-Targeted Antioxidants Protect against Ventilator-Induced Decreases in Diaphragm Muscle Protein Synthesis. PLoS ONE 2015, 10, e0137693. [Google Scholar] [CrossRef] [PubMed]
- O’Loghlen, A.; Perez-Morgado, M.I.; Salinas, M.; Martin, M.E. N-acetyl-cysteine abolishes hydrogen peroxide-induced modification of eukaryotic initiation factor 4F activity via distinct signalling pathways. Cell Signal. 2006, 18, 21–31. [Google Scholar] [CrossRef]
- Zhang, L.; Kimball, S.R.; Jefferson, L.S.; Shenberger, J.S. Hydrogen peroxide impairs insulin-stimulated assembly of mTORC1. Free Radic. Biol. Med. 2009, 46, 1500–1509. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Morton, A.B.; Ahn, B.; Smuder, A.J. Redox control of skeletal muscle atrophy. Free Radic. Biol. Med. 2016, 98, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smuder, A.J.; Sollanek, K.J.; Nelson, W.B.; Min, K.; Talbert, E.E.; Kavazis, A.N.; Hudson, M.B.; Sandri, M.; Szeto, H.H.; Powers, S.K. Crosstalk between autophagy and oxidative stress regulates proteolysis in the diaphragm during mechanical ventilation. Free Radic. Biol. Med. 2018, 115, 179–190. [Google Scholar] [CrossRef]
- Powell, S.R. The ubiquitin-proteasome system in cardiac physiology and pathology. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1–H19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grune, T.; Reinheckel, T.; Davies, K.J. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997, 11, 526–534. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef]
- Hyatt, H.W.; Ozdemir, M.; Yoshihara, T.; Nguyen, B.L.; Deminice, R.; Powers, S.K. Calpains play an essential role in mechanical ventilation-induced diaphragmatic weakness and mitochondrial dysfunction. Redox Biol. 2021, 38, 101802. [Google Scholar] [CrossRef]
- Whidden, M.A.; Smuder, A.J.; Wu, M.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J. Appl. Physiol. 2010, 108, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Smuder, A.J.; Kavazis, A.N.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic. Biol. Med. 2010, 49, 1152–1160. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, H.; Deminice, R.; Yoshihara, T.; Powers, S.K. Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: A review of the causes and effects. Arch. Biochem. Biophys. 2019, 662, 49–60. [Google Scholar] [CrossRef]
- Powers, S.K.; Wiggs, M.P.; Duarte, J.A.; Zergeroglu, A.M.; Demirel, H.A. Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E31–E39. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2015, 6, 422. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V. The Interplay between Mitochondrial Morphology and Myomitokines in Aging Sarcopenia. Int. J. Mol. Sci. 2020, 22, 91. [Google Scholar] [CrossRef]
- Carafoli, E.; Margreth, A.; Buffa, P. Early Biochemical Changes in Mitochondria from Denervated Muscle and Their Relation to the Onset of Atrophy. Exp. Mol. Pathol. 1964, 3, 171–181. [Google Scholar] [CrossRef]
- Kavazis, A.N.; Talbert, E.E.; Smuder, A.J.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic. Biol. Med. 2009, 46, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.; Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.K.; Hwang, M.H.; Szeto, H.H.; Kavazis, A.N.; et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015, 593, 2017–2036. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Hudson, M.B.; Nelson, W.B.; Talbert, E.E.; Min, K.; Szeto, H.H.; Kavazis, A.N.; Smuder, A.J. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 2011, 39, 1749–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbert, E.E.; Smuder, A.J.; Min, K.; Kwon, O.S.; Szeto, H.H.; Powers, S.K. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J. Appl. Physiol. 2013, 115, 529–538. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakellariou, G.K.; Pearson, T.; Lightfoot, A.P.; Nye, G.A.; Wells, N.; Giakoumaki, I.I.; Vasilaki, A.; Griffiths, R.D.; Jackson, M.J.; McArdle, A. Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci. Rep. 2016, 6, 33944. [Google Scholar] [CrossRef] [Green Version]
- Smuder, A.J.; Roberts, B.M.; Wiggs, M.P.; Kwon, O.S.; Yoo, J.K.; Christou, D.D.; Fuller, D.D.; Szeto, H.H.; Judge, A.R. Pharmacological targeting of mitochondrial function and reactive oxygen species production prevents colon 26 cancer-induced cardiorespiratory muscle weakness. Oncotarget 2020, 11, 3502–3514. [Google Scholar] [CrossRef]
- Powers, S.K.; Ozdemir, M.; Hyatt, H. Redox Control of Proteolysis During Inactivity-Induced Skeletal Muscle Atrophy. Antioxid. Redox. Signal. 2020, 33, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Espino, J.; Bejarano, I.; Lopez, J.J.; Rodriguez, A.B.; Pariente, J.A. Caspase-3 and -9 are activated in human myeloid HL-60 cells by calcium signal. Mol. Cell. Biochem. 2010, 333, 151–157. [Google Scholar] [CrossRef]
- Hyatt, H.W.; Powers, S.K. Disturbances in Calcium Homeostasis Promotes Skeletal Muscle Atrophy: Lessons From Ventilator-Induced Diaphragm Wasting. Front. Physiol. 2020, 11, 615351. [Google Scholar] [CrossRef] [PubMed]
- Aucello, M.; Dobrowolny, G.; Musaro, A. Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy 2009, 5, 527–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.P.; Chen, Y.; Li, A.S.; Reid, M.B. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am. J. Physiol. Cell Physiol. 2003, 285, C806–C812. [Google Scholar] [CrossRef] [Green Version]
- Alirezaei, M.; Marin, P.; Nairn, A.C.; Glowinski, J.; Premont, J. Inhibition of protein synthesis in cortical neurons during exposure to hydrogen peroxide. J. Neurochem. 2001, 76, 1080–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, F.H.; Sugden, P.H.; Clerk, A. Regulation of protein kinase B and 4E-BP1 by oxidative stress in cardiac myocytes. Circ. Res. 2000, 86, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Shenton, D.; Smirnova, J.B.; Selley, J.N.; Carroll, K.; Hubbard, S.J.; Pavitt, G.D.; Ashe, M.P.; Grant, C.M. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J. Biol. Chem. 2006, 281, 29011–29021. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Smuder, A.J.; Criswell, D.S. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid. Redox. Signal. 2011, 15, 2519–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzani, B.; Balage, M.; Venien, A.; Astruc, T.; Papet, I.; Dardevet, D.; Mosoni, L. Antioxidant supplementation restores defective leucine stimulation of protein synthesis in skeletal muscle from old rats. J. Nutr. 2008, 138, 2205–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloemberg, D.; Quadrilatero, J. Autophagy, apoptosis, and mitochondria: Molecular integration and physiological relevance in skeletal muscle. Am. J. Physiol. Cell Physiol. 2019, 317, C111–C130. [Google Scholar] [CrossRef]
- Max, S.R. Disuse atrophy of skeletal muscle: Loss of functional activity of mitochondria. Biochem. Biophys. Res. Commun. 1972, 46, 1394–1398. [Google Scholar] [CrossRef]
- Min, K.; Smuder, A.J.; Kwon, O.S.; Kavazis, A.N.; Szeto, H.H.; Powers, S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 2011, 111, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Thomson, D.M. The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. Int. J. Mol. Sci. 2018, 19, 3125. [Google Scholar] [CrossRef] [Green Version]
- Jewett, M.C.; Miller, M.L.; Chen, Y.; Swartz, J.R. Continued protein synthesis at low [ATP] and [GTP] enables cell adaptation during energy limitation. J. Bacteriol. 2009, 191, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanello, V.; Sandri, M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr. Hypertens. Rep. 2010, 12, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic. Biol. Med. 1999, 26, 463–471. [Google Scholar] [CrossRef]
- Levine, S.; Nguyen, T.; Taylor, N.; Friscia, M.E.; Budak, M.T.; Rothenberg, P.; Zhu, J.; Sachdeva, R.; Sonnad, S.; Kaiser, L.R.; et al. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N. Engl. J. Med. 2008, 358, 1327–1335. [Google Scholar] [CrossRef]
- Nelson, W.B.; Smuder, A.J.; Hudson, M.B.; Talbert, E.E.; Powers, S.K. Cross-talk between the calpain and caspase-3 proteolytic systems in the diaphragm during prolonged mechanical ventilation. Crit. Care Med. 2012, 40, 1857–1863. [Google Scholar] [CrossRef] [Green Version]
- Paddon-Jones, D.; Sheffield-Moore, M.; Cree, M.G.; Hewlings, S.J.; Aarsland, A.; Wolfe, R.R.; Ferrando, A.A. Atrophy and impaired muscle protein synthesis during prolonged inactivity and stress. J. Clin. Endocrinol. Metab. 2006, 91, 4836–4841. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.M.; Glover, E.I.; Rennie, M.J. Alterations of protein turnover underlying disuse atrophy in human skeletal muscle. J. Appl. Physiol. 2009, 107, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Shanely, R.A.; Van Gammeren, D.; Deruisseau, K.C.; Zergeroglu, A.M.; McKenzie, M.J.; Yarasheski, K.E.; Powers, S.K. Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am. J. Respir. Crit. Care Med. 2004, 170, 994–999. [Google Scholar] [CrossRef]
- Thomason, D.B.; Biggs, R.B.; Booth, F.W. Protein metabolism and beta-myosin heavy-chain mRNA in unweighted soleus muscle. Am. J. Physiol. 1989, 257, R300–R305. [Google Scholar] [CrossRef]
- Thomason, D.B.; Booth, F.W. Atrophy of the soleus muscle by hindlimb unweighting. J. Appl. Physiol. 1990, 68, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thomason, D.B.; Morrison, P.R.; Oganov, V.; Ilyina-Kakueva, E.; Booth, F.W.; Baldwin, K.M. Altered actin and myosin expression in muscle during exposure to microgravity. J. Appl. Physiol. 1992, 73, 90S–93S. [Google Scholar] [CrossRef]
- Kondo, H.; Miura, M.; Itokawa, Y. Oxidative stress in skeletal muscle atrophied by immobilization. Acta Physiol. Scand. 1991, 142, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Agten, A.; Maes, K.; Smuder, A.; Powers, S.K.; Decramer, M.; Gayan-Ramirez, G. N-Acetylcysteine protects the rat diaphragm from the decreased contractility associated with controlled mechanical ventilation. Crit. Care Med. 2011, 39, 777–782. [Google Scholar] [CrossRef]
- Appell, H.J.; Duarte, J.A.; Soares, J.M. Supplementation of vitamin E may attenuate skeletal muscle immobilization atrophy. Int. J. Sports Med. 1997, 18, 157–160. [Google Scholar] [PubMed]
- Betters, J.L.; Criswell, D.S.; Shanely, R.A.; Van Gammeren, D.; Falk, D.; Deruisseau, K.C.; Deering, M.; Yimlamai, T.; Powers, S.K. Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am. J. Respir. Crit. Care Med. 2004, 170, 1179–1184. [Google Scholar] [CrossRef] [Green Version]
- Laitano, O.; Ahn, B.; Patel, N.; Coblentz, P.D.; Smuder, A.J.; Yoo, J.K.; Christou, D.D.; Adhihetty, P.J.; Ferreira, L.F. Pharmacological targeting of mitochondrial reactive oxygen species counteracts diaphragm weakness in chronic heart failure. J. Appl. Physiol. 2016, 120, 733–742. [Google Scholar] [CrossRef] [Green Version]
- McClung, J.M.; Whidden, M.A.; Kavazis, A.N.; Falk, D.J.; Deruisseau, K.C.; Powers, S.K. Redox regulation of diaphragm proteolysis during mechanical ventilation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1608–R1617. [Google Scholar] [CrossRef] [Green Version]
- McClung, J.M.; Van Gammeren, D.; Whidden, M.A.; Falk, D.J.; Kavazis, A.N.; Hudson, M.B.; Gayan-Ramirez, G.; Decramer, M.; DeRuisseau, K.C.; Powers, S.K. Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit. Care Med. 2009, 37, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Whidden, M.A.; McClung, J.M.; Falk, D.J.; Hudson, M.B.; Smuder, A.J.; Nelson, W.B.; Powers, S.K. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J. Appl. Physiol. 2009, 106, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Abrigo, J.; Simon, F.; Cabrera, D.; Vilos, C.; Cabello-Verrugio, C. Mitochondrial Dysfunction in Skeletal Muscle Pathologies. Curr. Protein Pept. Sci. 2019, 20, 536–546. [Google Scholar] [CrossRef]
- Romanello, V.; Scalabrin, M.; Albiero, M.; Blaauw, B.; Scorrano, L.; Sandri, M. Inhibition of the Fission Machinery Mitigates OPA1 Impairment in Adult Skeletal Muscles. Cells 2019, 8, 597. [Google Scholar] [CrossRef] [Green Version]
- Supinski, G.S.; Schroder, E.A.; Callahan, L.A. Mitochondria and Critical Illness. Chest 2020, 157, 310–322. [Google Scholar] [CrossRef]
- Picard, M.; Jung, B.; Liang, F.; Azuelos, I.; Hussain, S.; Goldberg, P.; Godin, R.; Danialou, G.; Chaturvedi, R.; Rygiel, K.; et al. Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am. J. Respir. Crit. Care Med. 2012, 186, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Yamazaki, H.; Neufer, P.D. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J. Biol. Chem. 2007, 282, 31257–31266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Graham, Z.A.; Harlow, L.; Bauman, W.A.; Cardozo, C.P. Alterations in mitochondrial fission, fusion, and mitophagic protein expression in the gastrocnemius of mice after a sciatic nerve transection. Muscle Nerve 2018, 58, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.N.; Richards, B.J.; Slavin, M.; Hood, D.A. Exercise Is Muscle Mitochondrial Medicine. Exerc. Sport Sci. Rev. 2021, 49, 67–76. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell. Mol. Life Sci. 2021, 78, 1305–1328. [Google Scholar] [CrossRef]
- Matecki, S.; Dridi, H.; Jung, B.; Saint, N.; Reiken, S.R.; Scheuermann, V.; Mrozek, S.; Santulli, G.; Umanskaya, A.; Petrof, B.J.; et al. Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc. Natl. Acad. Sci. USA 2016, 113, 9069–9074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, E.; O’Rourke, B.; Maack, C. Mitochondria Do Not Survive Calcium Overload During Transplantation. Circ. Res. 2020, 126, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Boveris, A. Enhancement of hydrogen peroxide formation by protophores and ionophores in antimycin-supplemented mitochondria. Biochem. J. 1980, 188, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Castilho, R.F.; Kowaltowski, A.J.; Meinicke, A.R.; Bechara, E.J.; Vercesi, A.E. Permeabilization of the inner mitochondrial membrane by Ca2+ ions is stimulated by t-butyl hydroperoxide and mediated by reactive oxygen species generated by mitochondria. Free Radic. Biol. Med. 1995, 18, 479–486. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Castilho, R.F.; Vercesi, A.E. Ca(2+)-induced mitochondrial membrane permeabilization: Role of coenzyme Q redox state. Am. J. Physiol. 1995, 269, C141–C147. [Google Scholar] [CrossRef]
- Abid, H.; Ryan, Z.C.; Delmotte, P.; Sieck, G.C.; Lanza, I.R. Extramyocellular interleukin-6 influences skeletal muscle mitochondrial physiology through canonical JAK/STAT signaling pathways. FASEB J. 2020, 34, 14458–14472. [Google Scholar] [CrossRef]
- Smith, I.J.; Godinez, G.L.; Singh, B.K.; McCaughey, K.M.; Alcantara, R.R.; Gururaja, T.; Ho, M.S.; Nguyen, H.N.; Friera, A.M.; White, K.A.; et al. Inhibition of Janus kinase signaling during controlled mechanical ventilation prevents ventilation-induced diaphragm dysfunction. FASEB J. 2014, 28, 2790–2803. [Google Scholar] [CrossRef]
- Tang, H.; Smith, I.J.; Hussain, S.N.; Goldberg, P.; Lee, M.; Sugiarto, S.; Godinez, G.L.; Singh, B.K.; Payan, D.G.; Rando, T.A.; et al. The JAK-STAT pathway is critical in ventilator-induced diaphragm dysfunction. Mol. Med. 2015, 20, 579–589. [Google Scholar] [CrossRef]
- Ferreira, L.F.; Laitano, O. Regulation of NADPH oxidases in skeletal muscle. Free Radic. Biol. Med. 2016, 98, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Morton, A.B.; Hyatt, H.; Hinkley, M.J. The Renin-Angiotensin System and Skeletal Muscle. Exerc. Sport Sci. Rev. 2018, 46, 205–214. [Google Scholar] [CrossRef]
- Rosenberg, I.H. Sarcopenia: Origins and clinical relevance. J. Nutr. 1997, 127, 990S–991S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, R.N.; Koehler, K.M.; Gallagher, D.; Romero, L.; Heymsfield, S.B.; Ross, R.R.; Garry, P.J.; Lindeman, R.D. Epidemiology of sarcopenia among the elderly in New Mexico. Am. J. Epidemiol. 1998, 147, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Batsis, J.A.; Mackenzie, T.A.; Barre, L.K.; Lopez-Jimenez, F.; Bartels, S.J. Sarcopenia, sarcopenic obesity and mortality in older adults: Results from the National Health and Nutrition Examination Survey III. Eur. J. Clin. Nutr. 2014, 68, 1001–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, P.M.; Musci, R.V.; Hinkley, J.M.; Miller, B.F. Mitochondria as a Target for Mitigating Sarcopenia. Front. Physiol. 2018, 9, 1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.N.; Yoon, S.S. Sarcopenia: Neurological Point of View. J. Bone Metab. 2017, 24, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riuzzi, F.; Sorci, G.; Arcuri, C.; Giambanco, I.; Bellezza, I.; Minelli, A.; Donato, R. Cellular and molecular mechanisms of sarcopenia: The S100B perspective. J. Cachexia Sarcopenia Muscle 2018, 9, 1255–1268. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.J. Interactions between reactive oxygen species generated by contractile activity and aging in skeletal muscle? Antioxid. Redox Signal. 2013, 19, 804–812. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [Green Version]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.; Mansouri, A.; Van Remmen, H.; van der Meulen, J.H.; Larkin, L.; Richardson, A.G.; McArdle, A.; Faulkner, J.A.; Jackson, M.J. Free radical generation by skeletal muscle of adult and old mice: Effect of contractile activity. Aging Cell 2006, 5, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouspillou, G.; Bourdel-Marchasson, I.; Rouland, R.; Calmettes, G.; Biran, M.; Deschodt-Arsac, V.; Miraux, S.; Thiaudiere, E.; Pasdois, P.; Detaille, D.; et al. Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell 2014, 13, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A.; et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Chang, C.M.; Chi, C.W. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res. Rev. 2010, 9 (Suppl. 1), S47–S58. [Google Scholar] [CrossRef]
- Picard, M.; Turnbull, D.M. Linking the metabolic state and mitochondrial DNA in chronic disease, health, and aging. Diabetes 2013, 62, 672–678. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Beregi, E.; Regius, O.; Huttl, T.; Gobl, Z. Age-related changes in the skeletal muscle cells. Z Gerontol. 1988, 21, 83–86. [Google Scholar]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.J.; Vallee, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef]
- Joseph, A.M.; Adhihetty, P.J.; Wawrzyniak, N.R.; Wohlgemuth, S.E.; Picca, A.; Kujoth, G.C.; Prolla, T.A.; Leeuwenburgh, C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 2013, 8, e69327. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.D.; Duan, J.; Samuelson, A.T.; Gaffrey, M.J.; Merrihew, G.E.; Egertson, J.D.; Wang, L.; Bammler, T.K.; Moore, R.J.; White, C.C.; et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med. 2019, 134, 268–281. [Google Scholar] [CrossRef]
- Siegel, M.P.; Kruse, S.E.; Percival, J.M.; Goh, J.; White, C.C.; Hopkins, H.C.; Kavanagh, T.J.; Szeto, H.H.; Rabinovitch, P.S.; Marcinek, D.J. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 2013, 12, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, P.K.; Iliskovic, N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef]
- Smuder, A.J. Exercise stimulates beneficial adaptations to diminish doxorubicin-induced cellular toxicity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R662–R672. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Doroshow, J.H.; Davies, K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. 1986, 261, 3068–3074. [Google Scholar] [CrossRef]
- Gilliam, L.A.; Moylan, J.S.; Patterson, E.W.; Smith, J.D.; Wilson, A.S.; Rabbani, Z.; Reid, M.B. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2012, 302, C195–C202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J. Appl. Physiol. 2011, 111, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J. Appl. Physiol. 2011, 110, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalvo, R.N.; Doerr, V.; Min, K.; Szeto, H.H.; Smuder, A.J. Doxorubicin-induced oxidative stress differentially regulates proteolytic signaling in cardiac and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R227–R233. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Aggarwal, D.; Migrino, R.Q.; Joseph, J.; McAllister, D.; Konorev, E.A.; Antholine, W.E.; Zielonka, J.; Srinivasan, S.; Avadhani, N.G.; et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q. Biophys. J. 2009, 96, 1388–1398. [Google Scholar] [CrossRef] [Green Version]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Sun, L.; Quan, X.Q.; Yu, S. An Epidemiological Survey of Cachexia in Advanced Cancer Patients and Analysis on Its Diagnostic and Treatment Status. Nutr. Cancer 2015, 67, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Anker, M.S.; Holcomb, R.; Muscaritoli, M.; von Haehling, S.; Haverkamp, W.; Jatoi, A.; Morley, J.E.; Strasser, F.; Landmesser, U.; Coats, A.J.S.; et al. Orphan disease status of cancer cachexia in the USA and in the European Union: A systematic review. J. Cachexia Sarcopenia Muscle 2019, 10, 22–34. [Google Scholar] [CrossRef]
- Vagnildhaug, O.M.; Balstad, T.R.; Almberg, S.S.; Brunelli, C.; Knudsen, A.K.; Kaasa, S.; Thronaes, M.; Laird, B.; Solheim, T.S. A cross-sectional study examining the prevalence of cachexia and areas of unmet need in patients with cancer. Support. Care Cancer 2018, 26, 1871–1880. [Google Scholar] [CrossRef]
- Bozzetti, F.; Mariani, L. Defining and classifying cancer cachexia: A proposal by the SCRINIO Working Group. J. Parenter. Enter. Nutr. 2009, 33, 361–367. [Google Scholar] [CrossRef]
- Dolly, A.; Dumas, J.F.; Servais, S. Cancer cachexia and skeletal muscle atrophy in clinical studies: What do we really know? J. Cachexia Sarcopenia Muscle 2020, 11, 1413–1428. [Google Scholar] [CrossRef]
- Nosacka, R.L.; Delitto, A.E.; Delitto, D.; Patel, R.; Judge, S.M.; Trevino, J.G.; Judge, A.R. Distinct cachexia profiles in response to human pancreatic tumours in mouse limb and respiratory muscle. J. Cachexia Sarcopenia Muscle 2020, 11, 820–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johns, N.; Hatakeyama, S.; Stephens, N.A.; Degen, M.; Degen, S.; Frieauff, W.; Lambert, C.; Ross, J.A.; Roubenoff, R.; Glass, D.J.; et al. Clinical classification of cancer cachexia: Phenotypic correlates in human skeletal muscle. PLoS ONE 2014, 9, e83618. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Wang, X.; Gao, T.; Tian, H.; Zhou, D.; Zhang, L.; Li, G.; Wang, X. The autophagic-lysosomal and ubiquitin proteasome systems are simultaneously activated in the skeletal muscle of gastric cancer patients with cachexia. Am. J. Clin. Nutr. 2020, 111, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.M.; Nosacka, R.L.; Delitto, D.; Gerber, M.H.; Cameron, M.E.; Trevino, J.G.; Judge, A.R. Skeletal Muscle Fibrosis in Pancreatic Cancer Patients with Respect to Survival. JNCI Cancer Spectr. 2018, 2, pky043. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Caldwell, M.E.; Benson, C.A.; Lee, D.E.; Brown, J.L.; Washington, T.A.; Greene, N.P.; Wiggs, M.P. Mitochondrial Function and Protein Turnover in the Diaphragm are Altered in LLC Tumor Model of Cancer Cachexia. Int. J. Mol. Sci. 2020, 21, 7841. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.L.; Martignoni, M.E.; Bachmann, J.; Fechtner, K.; Friess, H.; Kinscherf, R.; Hildebrandt, W. Activity of the Akt-dependent anabolic and catabolic pathways in muscle and liver samples in cancer-related cachexia. J. Mol. Med. 2007, 85, 647–654. [Google Scholar] [CrossRef]
- Judge, S.M.; Wu, C.L.; Beharry, A.W.; Roberts, B.M.; Ferreira, L.F.; Kandarian, S.C.; Judge, A.R. Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 2014, 14, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padrao, A.I.; Oliveira, P.; Vitorino, R.; Colaco, B.; Pires, M.J.; Marquez, M.; Castellanos, E.; Neuparth, M.J.; Teixeira, C.; Costa, C.; et al. Bladder cancer-induced skeletal muscle wasting: Disclosing the role of mitochondria plasticity. Int. J. Biochem. Cell Biol. 2013, 45, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Oliveira, C.C.; Busquets, S.; Toledo, M.; Penna, F.; Paz Aylwin, M.; Sirisi, S.; Silva, A.P.; Orpi, M.; Garcia, A.; Sette, A.; et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: Altered energetic efficiency? Biochim. Biophys. Acta 2013, 1830, 2770–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shum, A.M.; Mahendradatta, T.; Taylor, R.J.; Painter, A.B.; Moore, M.M.; Tsoli, M.; Tan, T.C.; Clarke, S.J.; Robertson, G.R.; Polly, P. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging 2012, 4, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Castro, G.S.; Simoes, E.; Lima, J.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcantara, P.S.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 2019, 11, 1264. [Google Scholar] [CrossRef] [Green Version]
- White, J.P.; Baltgalvis, K.A.; Puppa, M.J.; Sato, S.; Baynes, J.W.; Carson, J.A. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R201–R211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzetti, E.; Lorenzi, M.; Landi, F.; Picca, A.; Rosa, F.; Tanganelli, F.; Galli, M.; Doglietto, G.B.; Pacelli, F.; Cesari, M.; et al. Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp. Gerontol. 2017, 87, 92–99. [Google Scholar] [CrossRef]
- Julienne, C.M.; Dumas, J.F.; Goupille, C.; Pinault, M.; Berri, C.; Collin, A.; Tesseraud, S.; Couet, C.; Servais, S. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J. Cachexia Sarcopenia Muscle 2012, 3, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fermoselle, C.; Garcia-Arumi, E.; Puig-Vilanova, E.; Andreu, A.L.; Urtreger, A.J.; de Kier Joffe, E.D.; Tejedor, A.; Puente-Maestu, L.; Barreiro, E. Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp. Physiol. 2013, 98, 1349–1365. [Google Scholar] [CrossRef] [Green Version]
- Mastrocola, R.; Reffo, P.; Penna, F.; Tomasinelli, C.E.; Boccuzzi, G.; Baccino, F.M.; Aragno, M.; Costelli, P. Muscle wasting in diabetic and in tumor-bearing rats: Role of oxidative stress. Free Radic. Biol. Med. 2008, 44, 584–593. [Google Scholar] [CrossRef]
- Fukawa, T.; Yan-Jiang, B.C.; Min-Wen, J.C.; Jun-Hao, E.T.; Huang, D.; Qian, C.N.; Ong, P.; Li, Z.; Chen, S.; Mak, S.Y.; et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 2016, 22, 666–671. [Google Scholar] [CrossRef]
- Brown, J.L.; Lawrence, M.M.; Ahn, B.; Kneis, P.; Piekarz, K.M.; Qaisar, R.; Ranjit, R.; Bian, J.; Pharaoh, G.; Brown, C.; et al. Cancer cachexia in a mouse model of oxidative stress. J. Cachexia Sarcopenia Muscle 2020, 11, 1688–1704. [Google Scholar] [CrossRef]
- Neyroud, D.; Nosacka, R.L.; Judge, A.R.; Hepple, R.T. Colon 26 adenocarcinoma (C26)-induced cancer cachexia impairs skeletal muscle mitochondrial function and content. J. Muscle Res. Cell Motil. 2019, 40, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pickrell, A.M.; Zimmers, T.A.; Moraes, C.T. Increase in muscle mitochondrial biogenesis does not prevent muscle loss but increased tumor size in a mouse model of acute cancer-induced cachexia. PLoS ONE 2012, 7, e33426. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef] [Green Version]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, C.E.; Bersten, A.D. Alterations in respiratory and limb muscle strength and size in patients with sepsis who are mechanically ventilated. Phys. Ther. 2014, 94, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Palakshappa, J.A.; Reilly, J.P.; Schweickert, W.D.; Anderson, B.J.; Khoury, V.; Shashaty, M.G.; Fitzgerald, D.; Forker, C.; Butler, K.; Ittner, C.A.; et al. Quantitative peripheral muscle ultrasound in sepsis: Muscle area superior to thickness. J. Crit. Care 2018, 47, 324–330. [Google Scholar] [CrossRef]
- Puthucheary, Z.A.; Rawal, J.; McPhail, M.; Connolly, B.; Ratnayake, G.; Chan, P.; Hopkinson, N.S.; Phadke, R.; Dew, T.; Sidhu, P.S.; et al. Acute skeletal muscle wasting in critical illness. JAMA 2013, 310, 1591–1600. [Google Scholar] [CrossRef] [Green Version]
- Jung, B.; Nougaret, S.; Conseil, M.; Coisel, Y.; Futier, E.; Chanques, G.; Molinari, N.; Lacampagne, A.; Matecki, S.; Jaber, S. Sepsis Is Associated with a Preferential Diaphragmatic Atrophy A Critically Ill Patient Study Using Tridimensional Computed Tomography. Anesthesiology 2014, 120, 1182–1191. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010, 304, 1787–1794. [Google Scholar] [CrossRef] [Green Version]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Svistunenko, D.A.; Davies, N.; Brealey, D.; Singer, M.; Cooper, C.E. Mitochondrial dysfunction in patients with severe sepsis: An EPR interrogation of individual respiratory chain components. Biochim. Biophys. Acta 2006, 1757, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, K.; Rooyackers, O. Mitochondrial function in sepsis: Respiratory versus leg muscle. Crit. Care Med. 2007, 35, S449–S453. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, K.; Hammarqvist, F.; Strigard, K.; Hultenby, K.; Ljungqvist, O.; Wernerman, J.; Rooyackers, O. Derangements in mitochondrial metabolism in intercostal and leg muscle of critically ill patients with sepsis-induced multiple organ failure. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1044–E1050. [Google Scholar] [CrossRef] [Green Version]
- Callahan, L.A.; Supinski, G.S. Sepsis induces diaphragm electron transport chain dysfunction and protein depletion. Am. J. Respir. Crit. Care Med. 2005, 172, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Callahan, L.A.; Supinski, G.S. Downregulation of diaphragm electron transport chain and glycolytic enzyme gene expression in sepsis. J. Appl. Physiol. 2005, 99, 1120–1126. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Mayaki, D.; Reynaud, O.; Broering, F.E.; Chaffer, T.J.; Hussain, S.N.A.; Gouspillou, G. Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting. Cells 2020, 9, 1454. [Google Scholar] [CrossRef]
- Fredriksson, K.; Tjader, I.; Keller, P.; Petrovic, N.; Ahlman, B.; Scheele, C.; Wernerman, J.; Timmons, J.A.; Rooyackers, O. Dysregulation of mitochondrial dynamics and the muscle transcriptome in ICU patients suffering from sepsis induced multiple organ failure. PLoS ONE 2008, 3, e3686. [Google Scholar] [CrossRef]
- Welty-Wolf, K.E.; Simonson, S.G.; Huang, Y.C.; Fracica, P.J.; Patterson, J.W.; Piantadosi, C.A. Ultrastructural changes in skeletal muscle mitochondria in gram-negative sepsis. Shock 1996, 5, 378–384. [Google Scholar] [CrossRef]
- Brealey, D.; Karyampudi, S.; Jacques, T.S.; Novelli, M.; Stidwill, R.; Taylor, V.; Smolenski, R.T.; Singer, M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R491–R497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peruchi, B.B.; Petronilho, F.; Rojas, H.A.; Constantino, L.; Mina, F.; Vuolo, F.; Cardoso, M.R.; Goncalves, C.L.; Rezin, G.T.; Streck, E.L.; et al. Skeletal muscle electron transport chain dysfunction after sepsis in rats. J. Surg. Res. 2011, 167, e333–e338. [Google Scholar] [CrossRef] [PubMed]
- Rocheteau, P.; Chatre, L.; Briand, D.; Mebarki, M.; Jouvion, G.; Bardon, J.; Crochemore, C.; Serrani, P.; Lecci, P.P.; Latil, M.; et al. Sepsis induces long-term metabolic and mitochondrial muscle stem cell dysfunction amenable by mesenchymal stem cell therapy. Nat. Commun. 2015, 6, 10145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeras, U.; Hall-Angeras, M.; Wagner, K.R.; James, H.; Hasselgren, P.O.; Fischer, J.E. Tissue metabolite levels in different types of skeletal muscle during sepsis. Metabolism 1991, 40, 1147–1151. [Google Scholar] [CrossRef]
- Supinski, G.S.; Wang, L.; Schroder, E.A.; Callahan, L.A.P. MitoTEMPOL, a mitochondrial targeted antioxidant, prevents sepsis-induced diaphragm dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L228–L238. [Google Scholar] [CrossRef]
- Callahan, L.A.; Supinski, G.S. Diaphragm and cardiac mitochondrial creatine kinases are impaired in sepsis. J. Appl. Physiol. 2007, 102, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Callahan, L.A.; Stofan, D.A.; Szweda, L.I.; Nethery, D.E.; Supinski, G.S. Free radicals alter maximal diaphragmatic mitochondrial oxygen consumption in endotoxin-induced sepsis. Free Radic. Biol. Med. 2001, 30, 129–138. [Google Scholar] [CrossRef]
- Javeshghani, D.; Magder, S.A.; Barreiro, E.; Quinn, M.T.; Hussain, S.N. Molecular characterization of a superoxide-generating NAD(P)H oxidase in the ventilatory muscles. Am. J. Respir. Crit. Care Med. 2002, 165, 412–418. [Google Scholar] [CrossRef]
- Clementi, E.; Brown, G.C.; Feelisch, M.; Moncada, S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. USA 1998, 95, 7631–7636. [Google Scholar] [CrossRef] [Green Version]
- Lopez, L.C.; Escames, G.; Tapias, V.; Utrilla, P.; Leon, J.; Acuna-Castroviejo, D. Identification of an inducible nitric oxide synthase in diaphragm mitochondria from septic mice: Its relation with mitochondrial dysfunction and prevention by melatonin. Int. J. Biochem. Cell Biol. 2006, 38, 267–278. [Google Scholar] [CrossRef]
- Boczkowski, J.; Lisdero, C.L.; Lanone, S.; Samb, A.; Carreras, M.C.; Boveris, A.; Aubier, M.; Poderoso, J.J. Endogenous peroxynitrite mediates mitochondrial dysfunction in rat diaphragm during endotoxemia. FASEB J. 1999, 13, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Supinski, G.S.; Wang, L.; Schroder, E.A.; Callahan, L.A.P. SS31, a mitochondrially targeted antioxidant, prevents sepsis-induced reductions in diaphragm strength and endurance. J. Appl. Physiol. 2020, 128, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Mofarrahi, M.; Sigala, I.; Guo, Y.; Godin, R.; Davis, E.C.; Petrof, B.; Sandri, M.; Burelle, Y.; Hussain, S.N. Autophagy and skeletal muscles in sepsis. PLoS ONE 2012, 7, e47265. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.N.; Kennedy, D.D.; Ghosh, I.R.; Misra, V.P.; Kiff, K.; Coakley, J.H.; Hinds, C.J. Persistent neuromuscular and neurophysiologic abnormalities in long-term survivors of prolonged critical illness. Crit. Care Med. 2003, 31, 1012–1016. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyatt, H.W.; Powers, S.K. Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity. Antioxidants 2021, 10, 588. https://doi.org/10.3390/antiox10040588
Hyatt HW, Powers SK. Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity. Antioxidants. 2021; 10(4):588. https://doi.org/10.3390/antiox10040588
Chicago/Turabian StyleHyatt, Hayden W., and Scott K. Powers. 2021. "Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity" Antioxidants 10, no. 4: 588. https://doi.org/10.3390/antiox10040588
APA StyleHyatt, H. W., & Powers, S. K. (2021). Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity. Antioxidants, 10(4), 588. https://doi.org/10.3390/antiox10040588