
Genomics of Viral Hepatitis-Associated Liver Tumors


Abstract
:1. Introduction
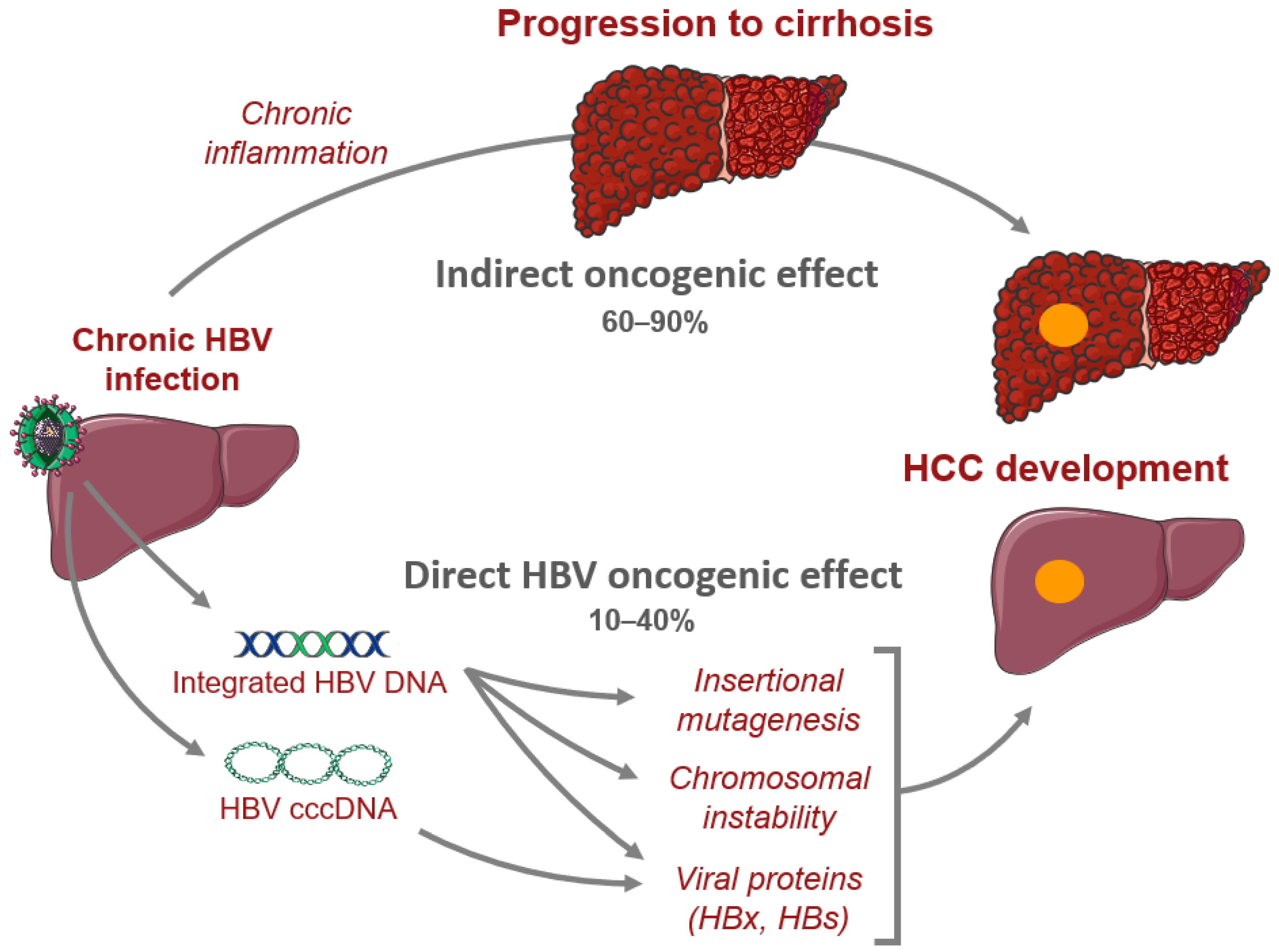
2. Genomics of Hepatitis B-Related HCC
2.1. HBV Structure and Cell Cycle
2.2. Genomic Landscape of HBV-Related HCC
2.3. HBV Oncogenic Proteins
2.4. HBV Integrations
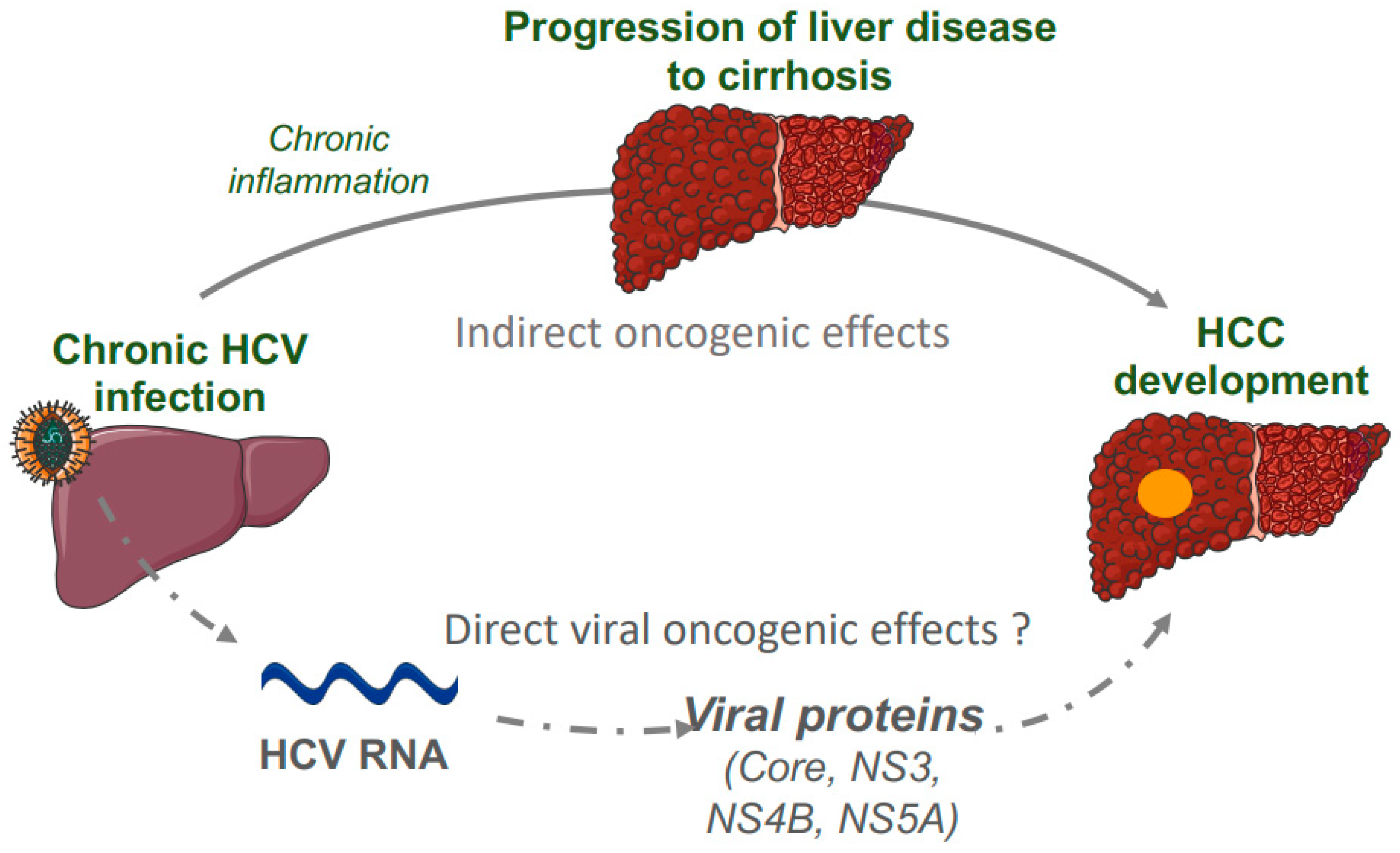
3. Genomics of Hepatitis C-Related HCC
3.1. HCV Structure and Cell Cycle
3.2. Genomic Landscape of HCV-Related HCC
3.3. HCV Oncogenic Proteins
4. Genomics of Hepatitis D-Related HCC
4.1. HDV Structure and Natural History of Infection
4.2. Genomic Landscape of HDV-Related HCC
5. Interactions with Exposures to Genotoxic Agents
5.1. HBV Infection and Aflatoxin B1 Exposure
5.2. HBV Infection and Aristolochic Acid Exposure
5.3. HDV Infection and Genotoxic Exposures
6. AAV as a New Player in Viral-Associated Liver Tumors
6.1. AAV Structure and Natural History of Infection in the Liver
6.2. AAV-Related Liver Carcinogenesis
7. Conclusions & Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global Hepatitis Report, 2017; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Block, T.M.; Alter, H.J.; London, W.T.; Bray, M. A historical perspective on the discovery and elucidation of the hepatitis B virus. Antivir. Res. 2016, 131, 109–123. [Google Scholar] [CrossRef]
- Blum, H.E. History and Global Burden of Viral Hepatitis. Dig. Dis. 2016, 34, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Razavi, H. Global Epidemiology of Viral Hepatitis. Gastroenterol. Clin. N. Am. 2020, 49, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Shirvani-Dastgerdi, E.; Schwartz, R.E.; Ploss, A. Hepatocarcinogenesis associated with hepatitis B, delta and C viruses. Curr. Opin. Virol. 2016, 20, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Nault, J.-C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, R.; Nault, J.-C.; Roberts, L.R.; Zucman-Rossi, J. Genomic Medicine and Implications for Hepatocellular Carcinoma Prevention and Therapy. Gastroenterology 2019, 156, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Ningarhari, M.; Caruso, S.; Hirsch, T.Z.; Bayard, Q.; Franconi, A.; Védie, A.-L.; Noblet, B.; Blanc, J.-F.; Amaddeo, G.; Ganne, N.; et al. Telomere length is key to hepatocellular carcinoma diversity and telomerase addiction is an actionable therapeutic target. J. Hepatol. 2021, 74, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, T.Z.; Negulescu, A.; Gupta, B.; Caruso, S.; Noblet, B.; Couchy, G.; Bayard, Q.; Meunier, L.; Morcrette, G.; Scoazec, J.-Y.; et al. BAP1 mutations define a homogeneous subgroup of hepatocellular carcinoma with fibrolamellar-like features and activated PKA. J. Hepatol. 2020, 72, 924–936. [Google Scholar] [CrossRef]
- Petruzziello, A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. TOVJ 2018, 12, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Romero, J.; Vargas, G.; García-Román, R. Occult HBV Infection: A Faceless Enemy in Liver Cancer Development. Viruses 2014, 6, 1590–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res. 2016, 2, 163. [Google Scholar] [CrossRef]
- Valaydon, Z.S.; Locarnini, S.A. The virological aspects of hepatitis B. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.; Shackel, N.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [Green Version]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaddeo, G.; Cao, Q.; Ladeiro, Y.; Imbeaud, S.; Nault, J.-C.; Jaoui, D.; Gaston Mathe, Y.; Laurent, C.; Laurent, A.; Bioulac-Sage, P.; et al. Integration of tumour and viral genomic characterisations in HBV-related hepatocellular carcinomas. Gut 2015, 64, 820–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Navas-Martin, S. Hepatitis B and C virus hepatocarcinogenesis: Lessons learned and future challenges. Cancer Lett. 2011, 305, 123–143. [Google Scholar] [CrossRef]
- Benhenda, S.; Cougot, D.; Buendia, M.-A.; Neuveut, C. Chapter 4 Hepatitis B Virus X Protein. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2009; Volume 103, pp. 75–109. [Google Scholar]
- Kew, M.C. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma: HBX and HCC. J. Gastroenterol. Hepatol. 2011, 26, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Brechot, C.; Pourcel, C.; Louise, A.; Rain, B.; Tiollais, P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature 1980, 286, 533–535. [Google Scholar] [CrossRef]
- Bréchot, C.; Gozuacik, D.; Murakami, Y.; Paterlini-Bréchot, P. Molecular bases for the development of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC). Semin. Cancer Biol. 2000, 10, 211–231. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, e02007-17. [Google Scholar] [CrossRef] [Green Version]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Cellular Genomic Sites of Hepatitis B Virus DNA Integration. Genes 2018, 9, 365. [Google Scholar] [CrossRef] [Green Version]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, S.T.; Jin, Y.; Liu, L.; Wang, J.; Babrzadeh, F.; Gharizadeh, B.; Ronaghi, M.; Toh, H.C.; Chow, P.K.-H.; Chung, A.Y.-F.; et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 2013, 34, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, C.; Lin, Y.; Ho, M.; Chen, D.; Chen, P.; Yeh, S. Androgen Receptor Enhances Hepatic Telomerase Reverse Transcriptase Gene Transcription After Hepatitis B Virus Integration or Point Mutation in Promoter Region. Hepatology 2019, 69, 498–512. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Sze, K.M.; Ho, D.W.; Chiu, Y.; Tsui, Y.; Chan, L.; Lee, J.M.; Chok, K.S.; Chan, A.C.; Tang, C.; Tang, V.W.; et al. HBV-TERT Promoter Integration Harnesses Host ELF4 Resulting in TERT Gene Transcription in Hepatocellular Carcinoma. Hepatology 2021, 73, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Zhang, L.; Qian, Z.; Zhu, X.; Zhu, G.; Chen, Y.; Xie, X.; Ye, Q.; Zang, J.; Ren, Z.; et al. Identification of HBV-MLL4 Integration and Its Molecular Basis in Chinese Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0123175. [Google Scholar] [CrossRef] [Green Version]
- Furuta, M.; Tanaka, H.; Shiraishi, Y.; Uchida, T.; Imamura, M.; Fujimoto, A.; Fujita, M.; Sasaki-Oku, A.; Maejima, K.; Nakano, K.; et al. Characterization of HBV integration patterns and timing in liver cancer and HBV-infected livers. Oncotarget 2018, 9, 25075–25088. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Fujimoto, A.; Furuta, M.; Tanaka, H.; Chiba, K.; Boroevich, K.A.; Abe, T.; Kawakami, Y.; Ueno, M.; Gotoh, K.; et al. Integrated Analysis of Whole Genome and Transcriptome Sequencing Reveals Diverse Transcriptomic Aberrations Driven by Somatic Genomic Changes in Liver Cancers. PLoS ONE 2014, 9, e114263. [Google Scholar] [CrossRef]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.-C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef] [Green Version]
- Péneau, C.; Imbeaud, S.; La Bella, T.; Hirsch, T.Z.; Caruso, S.; Calderaro, J.; Paradis, V.; Blanc, J.-F.; Letouzé, E.; Nault, J.-C.; et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut 2021, gutjnl-2020-323153. [Google Scholar] [CrossRef]
- Yoo, S.; Wang, W.; Wang, Q.; Fiel, M.I.; Lee, E.; Hiotis, S.P.; Zhu, J. A pilot systematic genomic comparison of recurrence risks of hepatitis B virus-associated hepatocellular carcinoma with low- and high-degree liver fibrosis. BMC Med. 2017, 15, 214. [Google Scholar] [CrossRef] [Green Version]
- Halgand, B.; Desterke, C.; Rivière, L.; Fallot, G.; Sebagh, M.; Calderaro, J.; Bioulac-Sage, P.; Neuveut, C.; Buendia, M.-A.; Samuel, D.; et al. Hepatitis B Virus Pregenomic RNA in Hepatocellular Carcinoma: A Nosological and Prognostic Determinant. Hepatology 2018, 67, 86–96. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Yuen, M.-F.; Chan, H.L.-Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [Green Version]
- Freitas, N.; Cunha, C.; Menne, S.; Gudima, S.O. Envelope Proteins Derived from Naturally Integrated Hepatitis B Virus DNA Support Assembly and Release of Infectious Hepatitis Delta Virus Particles. J. Virol. 2014, 88, 5742–5754. [Google Scholar] [CrossRef] [Green Version]
- Su, I.-J.; Wang, L.H.-C.; Hsieh, W.-C.; Wu, H.-C.; Teng, C.-F.; Tsai, H.-W.; Huang, W. The emerging role of hepatitis B virus Pre-S2 deletion mutant proteins in HBV tumorigenesis. J. Biomed. Sci. 2014, 21, 98. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, N.-F.; Lau, S.H.; Hu, L.; Xie, D.; Wu, J.; Yang, J.; Wang, Y.; Wu, M.-C.; Fung, J.; Bai, X.; et al. COOH-Terminal Truncated HBV X Protein Plays Key Role in Hepatocarcinogenesis. Clin. Cancer Res. 2008, 14, 5061–5068. [Google Scholar] [CrossRef] [Green Version]
- Sze, K.M.F.; Chu, G.K.Y.; Lee, J.M.F.; Ng, I.O.L. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by c-jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Andrisani, O.M.; Bouchard, M.J.; Lee, C.G.L.; Ou, J.-H.J.; Siddiqui, A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology 2015, 61, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-H.; Liu, X.; Yan, H.-X.; Li, W.-Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.-P.; Zhuang, X.-H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.-M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef]
- Ding, Q.; von Schaewen, M.; Ploss, A. The Impact of Hepatitis C Virus Entry on Viral Tropism. Cell Host Microbe 2014, 16, 562–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehermann, B. Hepatitis C virus versus innate and adaptive immune responses: A tale of coevolution and coexistence. J. Clin. Investig. 2009, 119, 1745–1754. [Google Scholar] [CrossRef] [Green Version]
- Atoom, A.M.; Taylor, N.G.A.; Russell, R.S. The elusive function of the hepatitis C virus p7 protein. Virology 2014, 462–463, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebouissou, S.; Nault, J.-C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshida, Y.; Nijman, S.M.B.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef] [Green Version]
- Bartosch, B.; Thimme, R.; Blum, H.E.; Zoulim, F. Hepatitis C virus-induced hepatocarcinogenesis. J. Hepatol. 2009, 51, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, A.S.; Seeff, L.B.; Morgan, T.R.; di Bisceglie, A.M.; Sterling, R.K.; Curto, T.M.; Everson, G.T.; Lindsay, K.L.; Lee, W.M.; Bonkovsky, H.L.; et al. Incidence of Hepatocellular Carcinoma and Associated Risk Factors in Hepatitis C-Related Advanced Liver Disease. Gastroenterology 2009, 136, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Yeh, M.M.; Daniel, H.D.-J.; Torbenson, M. Hepatitis C-associated hepatocellular carcinomas in non-cirrhotic livers. Mod. Pathol. 2010, 23, 276–283. [Google Scholar] [CrossRef]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, K.; Mochizuki, R.; Moriya, K.; Miyamoto, H.; Mori, Y.; Abe, T.; Murata, S.; Tanaka, K.; Miyamura, T.; Suzuki, T.; et al. Critical role of PA28 in hepatitis C virus-associated steatogenesis and hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 1661–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roingeard, P.; Hourioux, C. Hepatitis C virus core protein, lipid droplets and steatosis: HCV core, lipid droplets and steatosis. J. Viral Hepat. 2007, 15, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Zemel, R.; Gerechet, S.; Greif, H.; Bachmatove, L.; Birk, Y.; Golan-Goldhirsh, A.; Kunin, M.; Berdichevsky, Y.; Benhar, I.; Tur-Kaspa, R. Cell transformation induced by hepatitis C virus NS3 serine protease. J. Viral Hepat. 2001, 8, 96–102. [Google Scholar] [CrossRef]
- Smirnova, I.; Aksenov, N.; Vonsky, M.; Isaguliants, M. Different transformation pathways of murine fibroblast NIH 3T3 cells by hepatitis C virus core and NS3 proteins. Cell Biol. Int. 2006, 30, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Yang, J.M.; Min, M.-K. Hepatitis C Virus Nonstructural Protein NS4B Transforms NIH3T3 Cells in Cooperation with the Ha-ras Oncogene. Biochem. Biophys. Res. Commun. 2000, 267, 7. [Google Scholar] [CrossRef] [PubMed]
- Einav, S.; Sklan, E.H.; Moon, H.M.; Gehrig, E.; Liu, P.; Hao, Y.; Lowe, A.W.; Glenn, J.S. The nucleotide binding motif of hepatitis C virus NS4B can mediate cellular transformation and tumor formation without Ha-ras co-transfection. Hepatology 2008, 47, 827–835. [Google Scholar] [CrossRef]
- Park, C.-Y.; Jun, H.-J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C Virus Nonstructural 4B Protein Modulates Sterol Regulatory Element-binding Protein Signaling via the AKT Pathway. J. Biol. Chem. 2009, 284, 9237–9246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.-L.; Sheu, M.-L.; Yen, S.-H. Hepatitis C virus NS5A as a potential viral Bcl-2 homologue interacts with Bax and inhibits apoptosis in hepatocellular carcinoma. Int. J. Cancer 2003, 107, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, J.; Dallas, M.L.; Hughes, M.E.; Griffin, S.D.C.; Macdonald, A.; Peers, C.; Harris, M. Suppression of a pro-apoptotic K + channel as a mechanism for hepatitis C virus persistence. Proc. Natl. Acad. Sci. USA 2009, 106, 15903–15908. [Google Scholar] [CrossRef] [Green Version]
- Mentha, N.; Clément, S.; Negro, F.; Alfaiate, D. A review on hepatitis D: From virology to new therapies. J. Adv. Res. 2019, 17, 3–15. [Google Scholar] [CrossRef]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef]
- Sureau, C.; Guerra, B.; Lanford, R.E. Role of the large hepatitis B virus envelope protein in infectivity of the hepatitis delta virion. J. Virol. 1993, 67, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Bonino, F.; Heermann, K.H.; Rizzetto, M.; Gerlich, W.H. Hepatitis delta virus: Protein composition of delta antigen and its hepatitis B virus-derived envelope. J. Virol. 1986, 58, 945–950. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.M.C. The Molecular Biology of Hepatitis Delta Virus. Annu. Rev. Biochem. 1995, 64, 259–286. [Google Scholar] [CrossRef]
- Weiner, A.J.; Choo, Q.-L.; Wang, K.-S.; Govindarajan, S.; Houghton’, M. A Single Antigenomic Open Reading Frame of the Hepatitis Delta Virus Encodes the Epitope(s) of Both Hepatitis Delta Antigen Polypeptides p248 and p278. J. Virol. 1988, 62, 6. [Google Scholar] [CrossRef] [Green Version]
- Schaper, M.; Rodriguez-Frias, F.; Jardi, R.; Tabernero, D.; Homs, M.; Ruiz, G.; Quer, J.; Esteban, R.; Buti, M. Quantitative longitudinal evaluations of hepatitis delta virus RNA and hepatitis B virus DNA shows a dynamic, complex replicative profile in chronic hepatitis B and D. J. Hepatol. 2010, 52, 7. [Google Scholar] [CrossRef]
- Pollicino, T.; Raffa, G.; Santantonio, T.; Gaeta, G.B.; Iannello, G.; Alibrandi, A.; Squadrito, G.; Cacciola, I.; Calvi, C.; Colucci, G.; et al. Replicative and Transcriptional Activities of Hepatitis B Virus in Patients Coinfected with Hepatitis B and Hepatitis Delta Viruses. J. Virol. 2011, 85, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaiate, D. HDV RNA replication is associated with HBV repression and interferon-stimulated genes induction in super-infected hepatocytes. Antivir. Res. 2016, 136, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Giustina, G.; Christensen, E.; Pantalena, M.; Zagni, I.; Realdi, G.; Schalm, S.W. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. Gut 2000, 46, 420–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattovich, G.; Boscaro, S.; Noventa, F.; Pornaro, E.; Stenico, D.; Alberti, A.; Ruol, A.; Realdi, G. Influence of Hepatitis Delta Virus Infection on Progression to Cirrhosis in Chronic Hepatitis Type B. J. Infect. Dis. 1987, 155, 931–935. [Google Scholar] [CrossRef]
- Romeo, R.; Ninno, E.D.; Rumi, M.; Russo, A.; Sangiovanni, A.; Franchis, R.D.; Ronchi, G.; Colombo, M. A 28-Year Study of the Course of Hepatitis Δ Infection: A Risk Factor for Cirrhosis and Hepatocellular Carcinoma. Gastroenterology 2009, 136, 1629–1638. [Google Scholar] [CrossRef]
- Niro, G.A. Outcome of chronic delta hepatitis in Italy: A long-term cohort study. J. Hepatol. 2010, 53, 7. [Google Scholar] [CrossRef] [PubMed]
- Puigvehí, M.; Moctezuma-Velázquez, C.; Villanueva, A.; Llovet, J.M. The oncogenic role of hepatitis delta virus in hepatocellular carcinoma. JHEP Rep. 2019, 1, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, R.; Petruzziello, A.; Pecheur, E.I.; Facchetti, F.; Perbellini, R.; Galmozzi, E.; Khan, N.U.; Di Capua, L.; Sabatino, R.; Botti, G.; et al. Hepatitis delta virus and hepatocellular carcinoma: An update. Epidemiol. Infect. 2018, 146, 1612–1618. [Google Scholar] [CrossRef]
- Diaz, G.; Engle, R.E.; Tice, A.; Melis, M.; Montenegro, S.; Rodriguez-Canales, J.; Hanson, J.; Emmert-Buck, M.R.; Bock, K.W.; Moore, I.N.; et al. Molecular Signature and Mechanisms of Hepatitis D Virus–Associated Hepatocellular Carcinoma. Mol. Cancer Res. 2018, 16, 1406–1419. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Oidovsambuu, O.; Liu, P.; Grosely, R.; Elazar, M.; Winn, V.D.; Fram, B.; Boa, Z.; Dai, H.; Dashtseren, B.; et al. A novel quantitative microarray antibody capture (Q-MAC) assay identifies an extremely high HDV prevalence amongst HBV infected Mongolians. Hepatology 2017, 66, 1739–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candia, J.; Bayarsaikhan, E.; Tandon, M.; Budhu, A.; Forgues, M.; Tovuu, L.-O.; Tudev, U.; Lack, J.; Chao, A.; Chinburen, J.; et al. The genomic landscape of Mongolian hepatocellular carcinoma. Nat. Commun. 2020, 11, 4383. [Google Scholar] [CrossRef] [PubMed]
- Letouzé, E.; Shinde, J.; Renault, V.; Couchy, G.; Blanc, J.-F.; Tubacher, E.; Bayard, Q.; Bacq, D.; Meyer, V.; Semhoun, J.; et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 2017, 8, 1315. [Google Scholar] [CrossRef] [Green Version]
- Gouas, D.; Shi, H.; Hainaut, P. The aflatoxin-induced TP53 mutation at codon 249 (R249S): Biomarker of exposure, early detection and target for therapy. Cancer Lett. 2009, 286, 29–37. [Google Scholar] [CrossRef]
- Kew, M.C. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003, 23, 405–409. [Google Scholar] [CrossRef]
- Smela, M.E.; Currier, S.S.; Bailey, E.A.; Essigmann, J.M. The chemistry and biology of aflatoxin B1: From mutational spectrometry to carcinogenesis. Carcinogenesis 2001, 22, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; He, H.; Zang, M.; Wu, Q.; Zhao, H.; Lu, L.; Ma, P.; Zheng, H.; Wang, N.; Zhang, Y.; et al. Genetic Features of Aflatoxin-Associated Hepatocellular Carcinoma. Gastroenterology 2017, 153, 249–262.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlt, V.M. Aristolochic acid as a probable human cancer hazard in herbal remedies: A review. Mutagenesis 2002, 17, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, N.; Arlt, V.M.; Phillips, D.H.; Heflich, R.H.; Chen, T. DNA adduct formation and mutation induction by aristolochic acid in rat kidney and liver. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2006, 602, 83–91. [Google Scholar] [CrossRef]
- Poon, S.L.; Pang, S.-T.; McPherson, J.R.; Yu, W.; Huang, K.K.; Guan, P.; Weng, W.-H.; Siew, E.Y.; Liu, Y.; Heng, H.L.; et al. Genome-Wide Mutational Signatures of Aristolochic Acid and Its Application as a Screening Tool. Sci. Transl. Med. 2013, 5, 197ra101. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, Y.; Lin, M.; Lee, C.; Tsan, Y.; Lai, M.; Yang, H.; Ho, W.; Chen, P.; The Health Data Analysis in Taiwan (hDATa) Research Group. Herbal medicine containing aristolochic acid and the risk of hepatocellular carcinoma in patients with hepatitis B virus infection. Int. J. Cancer 2018, 143, 1578–1587. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Letouzé, E. Mutational Processes in Hepatocellular Carcinoma: The Story of Aristolochic Acid. Semin. Liver Dis. 2019, 39, 334–340. [Google Scholar] [CrossRef]
- Nault, J.-C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.-F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef]
- Gonçalves, M.A. Adeno-associated virus: From defective virus to effective vector. Virol. J. 2005, 2, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, B.R.; Chamberlain, J.S. Recombinant Adeno-associated Virus Transduction and Integration. Mol. Ther. 2008, 16, 1189–1199. [Google Scholar] [CrossRef]
- Smith, R.H. Adeno-associated virus integration: Virus versus vector. Gene Ther. 2008, 15, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-Associated Viruses Are Widely Disseminated in Human Tissues. JVI 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [Green Version]
- Zapatka, M.; Borozan, I.; Brewer, D.S.; Iskar, M.; Grundhoff, A.; Alawi, M.; Desai, N.; Sültmann, H.; Moch, H.; Cooper, C.S.; et al. The landscape of viral associations in human cancers. Nat. Genet. 2020, 52, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Tatsuno, K.; Midorikawa, Y.; Takayama, T.; Yamamoto, S.; Nagae, G.; Moriyama, M.; Nakagawa, H.; Koike, K.; Moriya, K.; Aburatani, H. Impact of AAV2 and Hepatitis B Virus Integration Into Genome on Development of Hepatocellular Carcinoma in Patients with Prior Hepatitis B Virus Infection. Clin. Cancer Res. 2019, 25, 6217–6227. [Google Scholar] [CrossRef] [PubMed]
- La Bella, T.; Imbeaud, S.; Peneau, C.; Mami, I.; Datta, S.; Bayard, Q.; Caruso, S.; Hirsch, T.Z.; Calderaro, J.; Morcrette, G.; et al. Adeno-associated virus in the liver: Natural history and consequences in tumour development. Gut 2020, 69, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Logan, G.J.; Dane, A.P.; Hallwirth, C.V.; Smyth, C.M.; Wilkie, E.E.; Amaya, A.K.; Zhu, E.; Khandekar, N.; Ginn, S.L.; Liao, S.H.Y.; et al. Identification of liver-specific enhancer–promoter activity in the 3′ untranslated region of the wild-type AAV2 genome. Nat. Genet. 2017, 49, 1267–1273. [Google Scholar] [CrossRef]
- Nguyen, G.N. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV Vector Integration Sites in Mouse Hepatocellular Carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.-C. Wild-type AAV Insertions in Hepatocellular Carcinoma Do Not Inform Debate Over Genotoxicity Risk of Vectorized AAV. Mol. Ther. 2016, 24, 660–661. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Virus | Genome | Number of Chronic Carriers | Development of Cirrhosis | Development of HCC | Oncogenic Mechanisms | Viral Oncoproteins |
|---|---|---|---|---|---|---|
| HAV | Single-stranded, positive-sense linear RNA virus | No chronic infection | No | No | -- | -- |
| HBV | Partially double-stranded, circular DNA virus | 257 millions | Yes | Yes | Chronic inflammation, oncoprotein, insertional mutagenesis | HBx, altered HBs or HBx |
| HCV | Single-stranded, positive-sense linear RNA virus | 71 millions | Yes | Yes | Chronic inflammation, oncoprotein | Core protein, NS3, NS4B, NS5A |
| HDV | Single-stranded, negative sense, closed circular RNA virus | 5% of HBV carriers | Yes | Yes | Chronic inflammation, oncoprotein | HDAg |
| HEV | Single-stranded, positive-sense linear RNA virus | Only in immuno compromized patient | Only in immuno compromized patient | Exceptionnal (immuno compromized patient) | -- | -- |
| AAV2 | Single-stranded DNA virus | 30–80% of positive serology in the general population 26% of AAV2 detected in liver | No | Yes (rare event occurring in normal liver) | Insertional mutagenesis | -- |
| Virus | HCC Features | Enriched Mutated Genes | Main Targeted Genes by Viral Integrations |
|---|---|---|---|
| Hepatitis B | Proliferative HCC, stem cell features, chromosomal instability | TP53 (R249S), IRF2 | TERT, MLL4, CCNE1 |
| Hepatitis C | Less agressive, hepatocyte-like features differentiated, chromosomal stability | TERT promoter, CTNNB1 | -- |
| Hepatitis D | Genetic instability | SPAT1 | -- |
| Adeno-Associated Virus type 2 | -- | PTEN, AXIN1 | CCNA2, CCNE1, GLI1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Péneau, C.; Zucman-Rossi, J.; Nault, J.-C. Genomics of Viral Hepatitis-Associated Liver Tumors. J. Clin. Med. 2021, 10, 1827. https://doi.org/10.3390/jcm10091827
Péneau C, Zucman-Rossi J, Nault J-C. Genomics of Viral Hepatitis-Associated Liver Tumors. Journal of Clinical Medicine. 2021; 10(9):1827. https://doi.org/10.3390/jcm10091827
Chicago/Turabian StylePéneau, Camille, Jessica Zucman-Rossi, and Jean-Charles Nault. 2021. "Genomics of Viral Hepatitis-Associated Liver Tumors" Journal of Clinical Medicine 10, no. 9: 1827. https://doi.org/10.3390/jcm10091827
APA StylePéneau, C., Zucman-Rossi, J., & Nault, J.-C. (2021). Genomics of Viral Hepatitis-Associated Liver Tumors. Journal of Clinical Medicine, 10(9), 1827. https://doi.org/10.3390/jcm10091827