
Influence of N-Methylation and Conformation on Almiramide Anti-Leishmanial Activity


 and
and 
Abstract
:1. Introduction
2. Results and Discussion
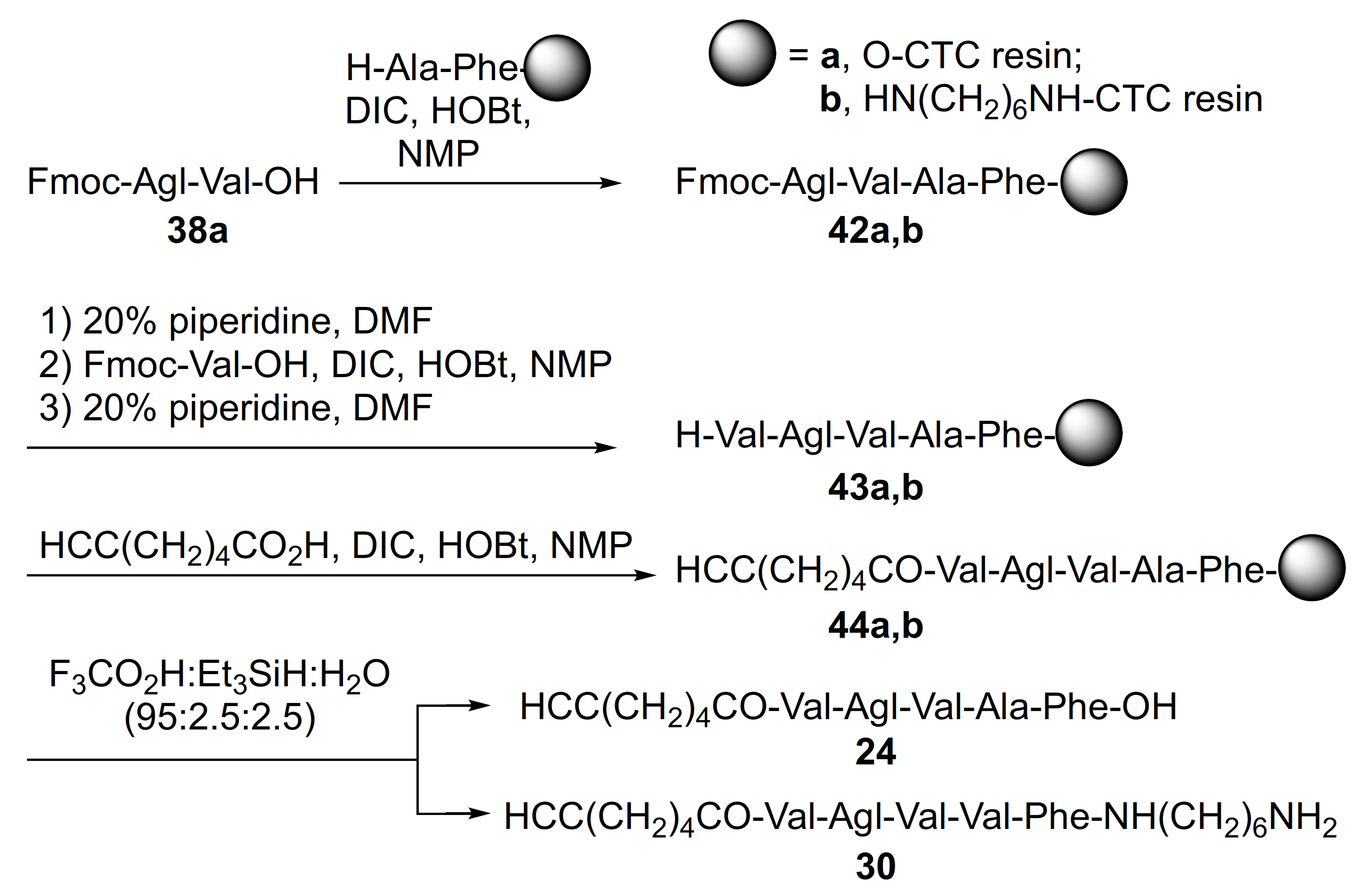
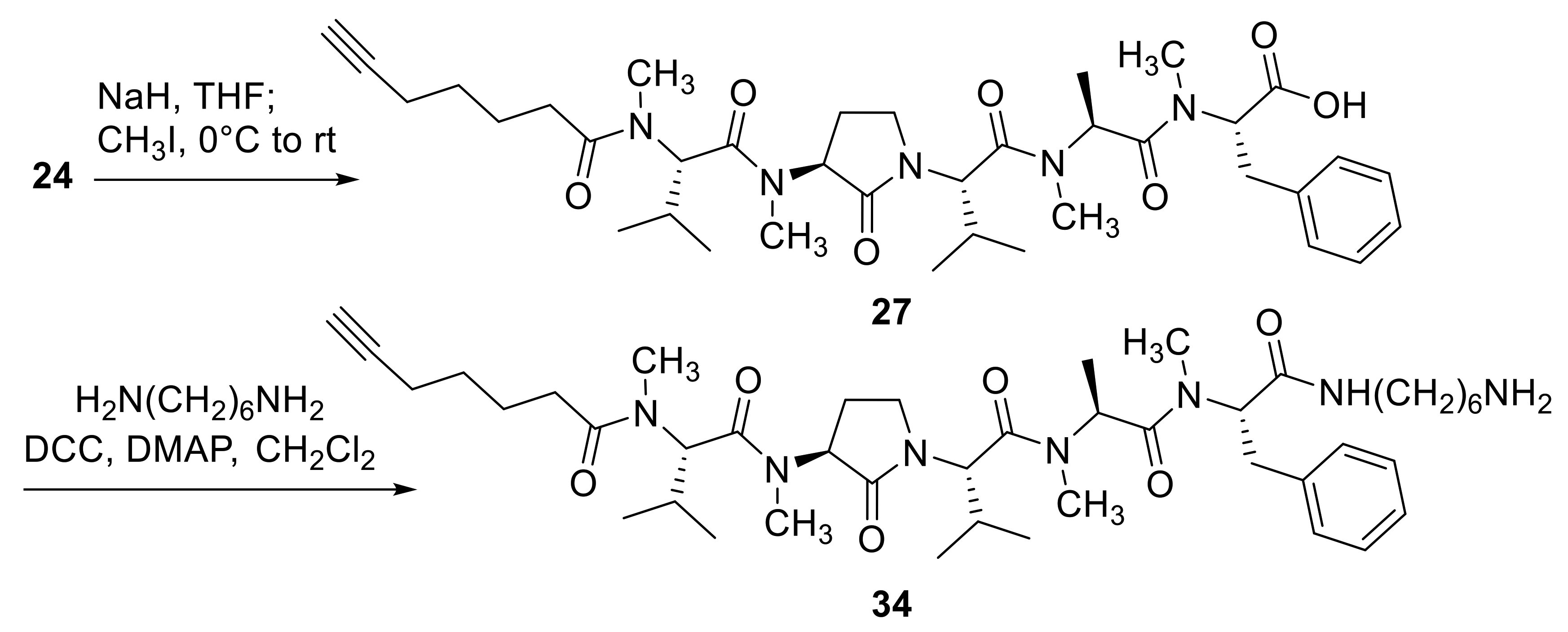
2.1. Chemistry
2.2. Bioactivity
3. Discussion
4. Materials and Methods
4.1. Experimental Section
4.1.1. Leishmania Cultures and Antileishmanial Activity Determination
4.1.2. LM-1 Macrophages and Cytotoxicity Determination
4.2. Materials












5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug. Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Prada, C.; Douanne, N.; Minguez-Menendez, A.; Pena, J.; Tunes, L.G.; Pires, D.E.; Monte-Neto, R.L. Repurposed Molecules: A New Hope in Tackling Neglected Infectious Diseases. In In Silico Drug Design; Roy, K., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 119–160. [Google Scholar]
- Pramanik, P.K.; Alam, M.N.; Chowdhury, D.R.; Chakraborti, T. Drug resistance in protozoan parasites: An incessant wrestle for survival. J. Glob. Antimicrob. Resist. 2019, 18, 1–11. [Google Scholar] [CrossRef]
- Lupi, O.; Bartlett, B.L.; Haugen, R.N.; Dy, L.C.; Sethi, A.; Klaus, S.N.; Pinto, J.M.; Bravo, F.; Tyring, S.K. Tropical dermatology: Tropical diseases caused by protozoa. J. Am. Acad. Dermatol. 2009, 60, 897–925. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2019, 392, 951–970. [Google Scholar] [CrossRef]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; Team, W.L.C. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Balaña-Fouce, R.; Pertejo, M.Y.P.; Domínguez-Asenjo, B.; Gutiérrez-Corbo, C.; Reguera, R.M. Walking a tightrope: Drug discovery in visceral leishmaniasis. Drug Discov. Today 2019, 24, 1209–1216. [Google Scholar] [CrossRef]
- Mehdi, D.S. The effect of visceral leishmaniasis on some liver enzyme and blood parameter. J. Thiqar Univ. 2008, 4, 2–5. [Google Scholar]
- Bilgic-Temel, A.; Murrell, D.F.; Uzun, S. Cutaneous leishmaniasis: A neglected disfiguring disease for women. Int. J. Womens Dermatol. 2019, 5, 158–165. [Google Scholar] [CrossRef]
- Strazzulla, A.; Cocuzza, S.; Pinzone, M.R.; Postorino, M.C.; Cosentino, S.; Serra, A.; Cacopardo, B.; Nunnari, G. Mucosal leishmaniasis: An underestimated presentation of a neglected disease. Biochem. Biophys. Res. Commun. 2013, 2013, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lindoso, J.A.L.; Moreira, C.H.V.; Cunha, M.A.; Queiroz, I.T. Visceral leishmaniasis and HIV coinfection: Current perspectives. HIV AIDS 2018, 10, 193. [Google Scholar] [CrossRef] [Green Version]
- Uliana, S.R.B.; Trinconi, C.T.; Coelho, A.C. Chemotherapy of leishmaniasis: Present challenges. Parasitology 2018, 145, 464. [Google Scholar] [CrossRef]
- Sneader, W. Chemical Medicines. In Drug Discovery: A History; John Wiley and Sons: Hoboken, NJ, USA, 2005; pp. 57–59. [Google Scholar]
- Sundar, S.; More, D.K.; Singh, M.K.; Singh, V.P.; Sharma, S.; Makharia, A.; Kumar, P.C.; Murray, H.W. Failure of pentavalent antimony in visceral leishmaniasis in India: Report from the center of the Indian epidemic. Clin. Infect. Dis. 2000, 31, 1104–1107. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, S.A.; Godoy, J.T.; Paiva, L.; Dillon, N.L.; Lacaz, C.D.S. The treatment of American (mucocutaneous) leishmaniasis with amphotericin B. Arch. Dermatol. 1960, 82, 627–635. [Google Scholar] [CrossRef]
- Sundar, S.; Jha, T.; Thakur, C.; Engel, J.; Sindermann, H.; Fischer, C.; Junge, K.; Bryceson, A.; Berman, J. Oral miltefosine for Indian visceral leishmaniasis. N. Eng. J. Med. 2002, 347, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Jha, T.; Thakur, C.P.; Sinha, P.K.; Bhattacharya, S.K. Injectable paromomycin for visceral leishmaniasis in India. N. Eng. J. Med. 2007, 356, 2571–2581. [Google Scholar] [CrossRef] [Green Version]
- Hailu, A.; Musa, A.; Wasunna, M.; Balasegaram, M.; Yifru, S.; Mengistu, G.; Hurissa, Z.; Hailu, W.; Weldegebreal, T.; Tesfaye, S. Geographical variation in the response of visceral leishmaniasis to paromomycin in East Africa: A multicentre, open-label, randomized trial. PLoS Negl. Trop. Dis. 2010, 4, e709. [Google Scholar] [CrossRef] [Green Version]
- Brindha, J.; Balamurali, M.M.; Chanda, K. An Overview on the Therapeutics of Neglected Infectious Diseases—Leishmaniasis and Chagas Diseases. Front. Chem. 2021, 9, 1–19. [Google Scholar] [CrossRef]
- Silva, C.F.; Pinto, D.C.; Fernandes, P.A.; Silva, A.M. Evolution of chromone-like compounds as potential antileishmanial agents, through the 21st century. Expert Opin. Drug Discov. 2020, 15, 1425–1439. [Google Scholar] [CrossRef]
- Ortalli, M.; Varani, S.; Cimato, G.; Veronesi, R.; Quintavalla, A.; Lombardo, M.; Monari, M.; Trombini, C. Evaluation of the pharmacophoric role of the O–O bond in synthetic antileishmanial compounds: Comparison between 1, 2-dioxanes and tetrahydropyrans. J. Med. Chem. 2020, 63, 13140–13158. [Google Scholar] [CrossRef]
- Shalev, M.; Rozenberg, H.; Smolkin, B.; Nasereddin, A.; Kopelyanskiy, D.; Belakhov, V.; Schrepfer, T.; Schacht, J.; Jaffe, C.L.; Adir, N. Structural basis for selective targeting of leishmanial ribosomes: Aminoglycoside derivatives as promising therapeutics. Nucleic Acids Res. 2015, 43, 8601–8613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagle, A.; Biggart, A.; Be, C.; Srinivas, H.; Hein, A.; Caridha, D.; Sciotti, R.J.; Pybus, B.; Kreishman-Deitrick, M.; Bursulaya, B. Discovery and characterization of clinical candidate LXE408 as a kinetoplastid-selective proteasome inhibitor for the treatment of leishmaniases. J. Med. Chem. 2020, 63, 10773–10781. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.M.; Lopez, D.; Vesely, B.A.; Della Togna, G.; Gerwick, W.H.; Kyle, D.E.; Linington, R.G. Almiramides A− C: Discovery and development of a new class of leishmaniasis lead compounds. J. Med. Chem. 2010, 53, 4187–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, L.M.; Knudsen, G.M.; Helbig, C.; De Muylder, G.; Mascuch, S.M.; Mackey, Z.B.; Gerwick, L.; Clayton, C.; McKerrow, J.H.; Linington, R.G. Examination of the mode of action of the almiramide family of natural products against the kinetoplastid parasite Trypanosoma brucei. J. Nat. Prod. 2013, 76, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Kalel, V.C.; Mäser, P.; Sattler, M.; Erdmann, R.; Popowicz, G.M. Come, sweet death: Targeting glycosomal protein import for antitrypanosomal drug development. Curr. Opin. Microbiol. 2018, 46, 116–122. [Google Scholar] [CrossRef]
- Michels, P.A.; Bringaud, F.; Herman, M.; Hannaert, V. Metabolic functions of glycosomes in trypanosomatids. Biochim. Biophys. Acta 2006, 1763, 1463–1477. [Google Scholar] [CrossRef]
- Dawidowski, M.; Emmanouilidis, L.; Kalel, V.; Tripsianes, K.; Schorpp, K.; Hadian, K.; Kaiser, M.; Mäser, P.; Kolonko, M.; Tanghe, S. Inhibitors of PEX14 disrupt protein import into glycosomes and kill Trypanosoma parasites. Science 2017, 355, 1416–1420. [Google Scholar] [CrossRef]
- Dawidowski, M.; Kalel, V.C.; Napolitano, V.; Fino, R.; Schorpp, K.; Emmanouilidis, L.; Lenhart, D.; Ostertag, M.; Kaiser, M.; Kolonko, M. Structure–activity relationship in pyrazolo [4, 3-c] pyridines, first inhibitors of PEX14–PEX5 protein–protein interaction with trypanocidal activity. J. Med. Chem. 2019, 63, 847–879. [Google Scholar] [CrossRef]
- Quintana, J.; Bayona, L.M.; Castellanos, L.; Puyana, M.; Camargo, P.; Aristizábal, F.; Edwards, C.; Tabudravu, J.N.; Jaspars, M.; Ramos, F.A. Almiramide D, cytotoxic peptide from the marine cyanobacterium Oscillatoria nigroviridis. Bioorg. Med. Chem. 2014, 22, 6789–6795. [Google Scholar] [CrossRef]
- Das, D.; Khan, H.P.; Shivahare, R.; Gupta, S.; Sarkar, J.; Siddiqui, M.I.; Ampapathi, R.S.; Chakraborty, T.K. Synthesis, SAR and biological studies of sugar amino acid-based almiramide analogues: N-methylation leads the way. Org. Biomol. Chem. 2017, 15, 3337–3352. [Google Scholar] [CrossRef]
- Freidinger, R.M.; Hinkle, J.S.; Perlow, D.S. Synthesis of 9-fluorenylmethyloxycarbonyl-protected N-alkyl amino acids by reduction of oxazolidinones. J. Org. Chem. 1983, 48, 77–81. [Google Scholar] [CrossRef]
- Freidinger, R.M.; Veber, D.F.; Perlow, D.S.; Saperstein, R. Bioactive conformation of luteinizing hormone-releasing hormone: Evidence from a conformationally constrained analog. Science 1980, 210, 656–658. [Google Scholar] [CrossRef]
- St-Cyr, D.J.; García-Ramos, Y.; Doan, N.-D.; Lubell, W.D. Aminolactam, N-aminoimidazolone, and N-aminoimdazolidinone peptide mimics. In Peptidomimetics I.; Lubell, W.D., Ed.; Springer: Berlin, Germany, 2017; pp. 125–175. [Google Scholar]
- Poupart, J.; Hamdane, Y.; Lubell, W.D. Synthesis of enantiomerically enriched 4, 5-disubstituted N-aminoimidazol-2-one (Nai) peptide turn mimics. Can. J. Chem. 2020, 98, 278–284. [Google Scholar] [CrossRef]
- Geranurimi, A.; Cheng, C.W.; Quiniou, C.; Zhu, T.; Hou, X.; Rivera, J.C.; St-Cyr, D.J.; Beauregard, K.; Bernard-Gauthier, V.; Chemtob, S.; et al. Probing anti-inflammatory properties independent of NF-κB through conformational constraint of peptide-based interleukin-1 receptor biased ligands. Front. Chem. 2019, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Hamdane, Y.; Chauhan, P.S.; Vutla, S.; Mulumba, M.; Ong, H.; Lubell, W.D. 5-Substituted N-aminoimidazolone peptide mimic synthesis by organocatalyzed reactions of azopeptides and use in conformational analysis of biologically active backbone and side chain topology. Org. Lett. 2021, 23, 3491–3495. [Google Scholar] [CrossRef]
- Lubell, W.D.; Blankenship, J.W.; Fridkin, G.; Kaul, R. Product Class 11: Peptides. In Three Carbon-Heteroatom Bonds: Amides and Derivatives, Peptides, Lactams; Weinreb, S., Ed.; Georg Thieme Verlag KG: New York, NY, USA, 2005; pp. 713–810. [Google Scholar]
- Miller, S.C.; Scanlan, T.S. Site-selective N-methylation of peptides on solid support. J. Am. Chem. Soc. 1997, 119, 2301–2302. [Google Scholar] [CrossRef]
- Quiñones, W.; Acosta, H.; Gonçalves, C.S.; Motta, M.C.M.; Gualdrón-López, M.; Michels, P.A. Structure, properties, and function of glycosomes in Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 2020, 10, 25. [Google Scholar] [CrossRef]
- Gualdron-Lopez, M.; Brennand, A.; Avilan, L.; Michels, P.A. Translocation of solutes and proteins across the glycosomal membrane of trypanosomes; possibilities and limitations for targeting with trypanocidal drugs. Parasitology 2013, 140, 1. [Google Scholar] [CrossRef]
- Mohapatra, S. Drug resistance in leishmaniasis: Newer developments. Trop. Parasitol. 2014, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Mbongo, N.; Loiseau, P.M.; Billion, M.A.; Robert-Gero, M. Mechanism of amphotericin B resistance in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 1998, 42, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Katakai, R.; Toda, F.; Uno, K.; Iwakura, Y.; Oya, M. Conformation of sequential polypeptides of L-valine in solution. Chem. Lett. 1973, 2, 763–768. [Google Scholar] [CrossRef]
- Teixidó, M.; Albericio, F.; Giralt, E. Solid-phase synthesis and characterization of N-methyl-rich peptides. J. Pept. Res. 2005, 65, 153–166. [Google Scholar] [CrossRef]
- Revilla-López, G.; Rodríguez-Ropero, F.; Curcó, D.; Torras, J.; Isabel Calaza, M.; Zanuy, D.; Jiménez, A.I.; Cativiela, C.; Nussinov, R.; Alemán, C. Integrating the intrinsic conformational preferences of noncoded α-amino acids modified at the peptide bond into the noncoded amino acids database. Proteins 2011, 79, 1841–1852. [Google Scholar] [CrossRef] [Green Version]
- Doedens, L.; Opperer, F.; Cai, M.; Beck, J.G.; Dedek, M.; Palmer, E.; Hruby, V.J.; Kessler, H. Multiple N-methylation of MT-II backbone amide bonds leads to melanocortin receptor subtype hMC1R selectivity: Pharmacological and conformational studies. J. Am. Chem. Soc. 2010, 132, 8115–8128. [Google Scholar] [CrossRef] [Green Version]
- Merlino, F.; Billard, É.; Yousif, A.M.; Di Maro, S.; Brancaccio, D.; Abate, L.; Carotenuto, A.; Bellavita, R.; d’Emmanuele di Villa Bianca, R.; Santicioli, P. Functional selectivity revealed by N-methylation scanning of human urotensin II and related peptides. J. Med. Chem. 2019, 62, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, M.G.; Moya, C.G.; Pérez, C.S.; Porzel, A.; Wessjohann, L.A.; Rivera, D.G. Improved stability and tunable functionalization of parallel β-sheets via multicomponent N-alkylation of the turn moiety. Angew. Chem. Int. Ed. 2020, 59, 259–263. [Google Scholar] [CrossRef] [PubMed]
- St-Cyr, D.J.; Maris, T.; Lubell, W.D. Crystal-state structure analysis of β-hydroxy-γ-lactam constrained Ser/Thr peptidomimetics. Heterocycles 2010, 82, 729–737. [Google Scholar]
- Leprohon, P.; Legare, D.; Raymond, F.; Madore, E.; Hardiman, G.; Corbeil, J.; Ouellette, M. Gene expression modulation is associated with gene amplification, supernumerary chromosomes and chromosome loss in antimony-resistant Leishmania infantum. Nucleic Acids Res. 2009, 37, 1387–1399. [Google Scholar] [CrossRef] [Green Version]
- Brotherton, M.-C.; Bourassa, S.; Leprohon, P.; Légaré, D.; Poirier, G.G.; Droit, A.; Ouellette, M. Proteomic and genomic analyses of antimony resistant Leishmania infantum mutant. PLoS ONE 2013, 8, e81899. [Google Scholar] [CrossRef] [Green Version]
- El Fadili, K.; Messier, N.; Leprohon, P.; Roy, G.; Guimond, C.; Trudel, N.; Saravia, N.G.; Papadopoulou, B.; Légaré, D.; Ouellette, M. Role of the ABC transporter MRPA (PGPA) in antimony resistance in Leishmania infantum axenic and intracellular amastigotes. Antimicrob. Agents Chemother. 2005, 49, 1988–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brotherton, M.-C.; Bourassa, S.; Légaré, D.; Poirier, G.G.; Droit, A.; Ouellette, M. Quantitative proteomic analysis of amphotericin B resistance in Leishmania infantum. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 126–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Prada, C.; Vincent, I.M.; Brotherton, M.-C.; Roberts, M.; Roy, G.; Rivas, L.; Leprohon, P.; Smith, T.K.; Ouellette, M. Different mutations in a P-type ATPase transporter in Leishmania parasites are associated with cross-resistance to two leading drugs by distinct mechanisms. PLoS Negl. Trop. Dis. 2016, 10, e0005171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, W.; Leprohon, P.; Ouellette, M. Tolerance to drug-induced cell death favours the acquisition of multidrug resistance in Leishmania. Cell Death Dis. 2011, 2, e201. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
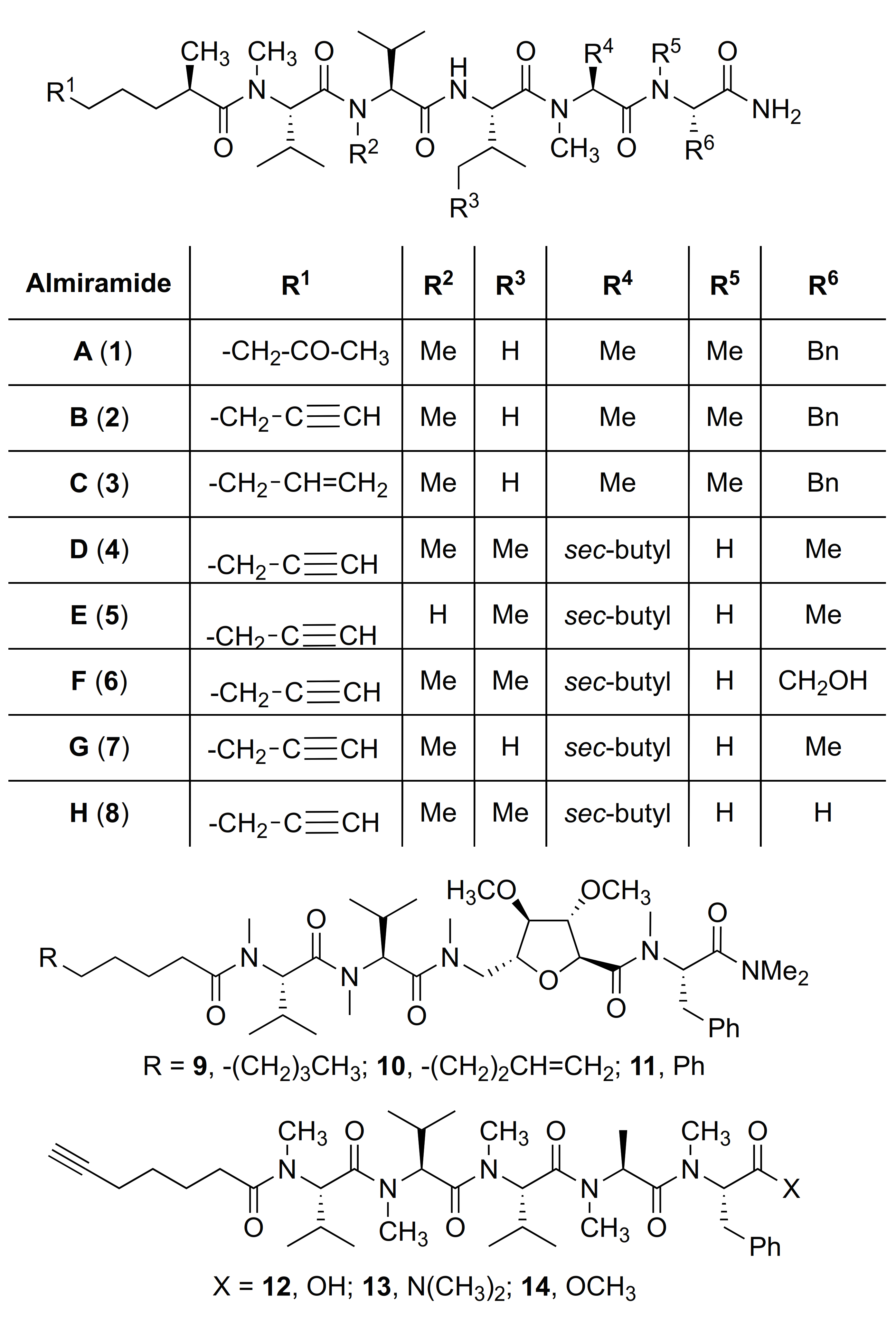
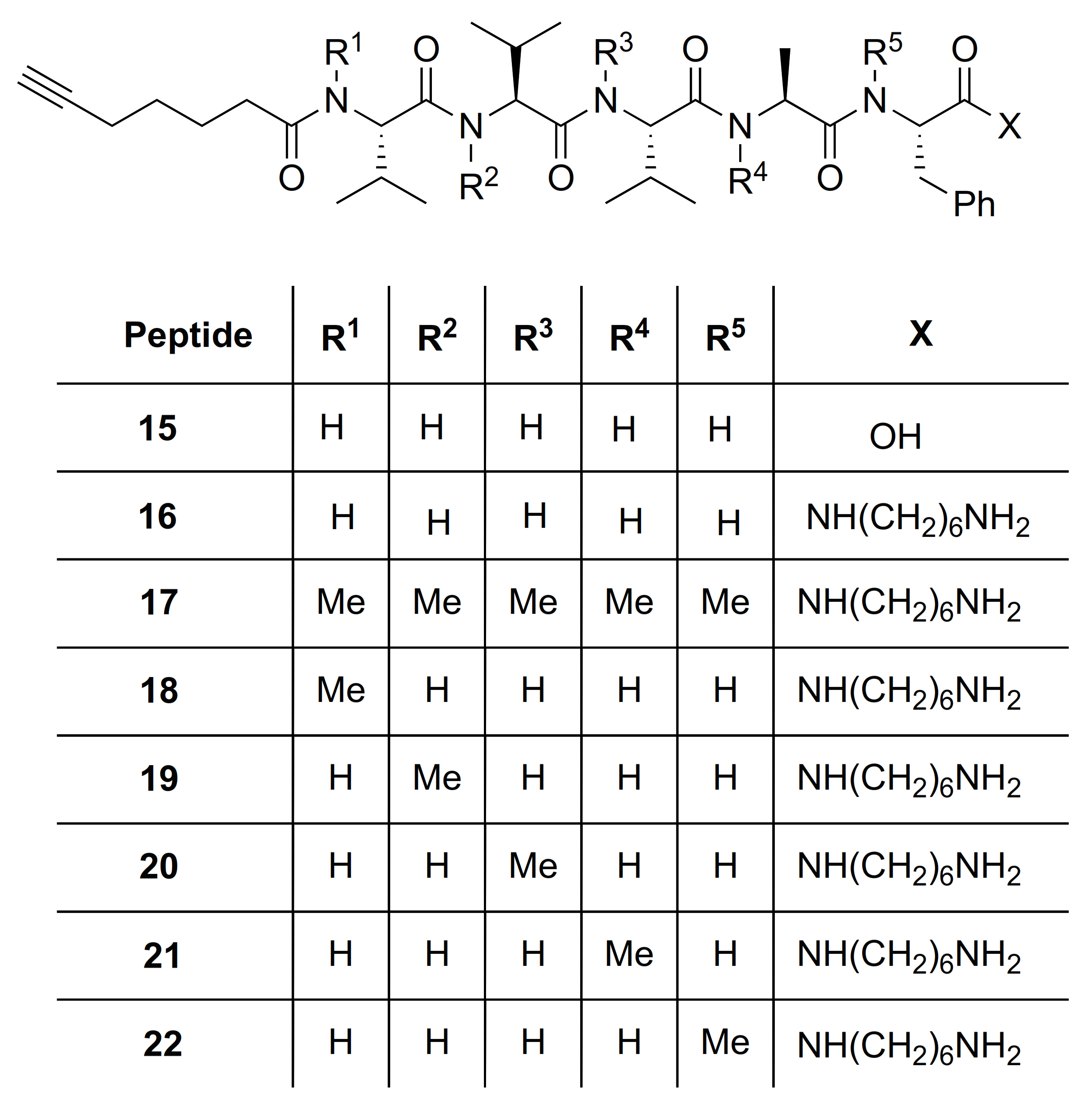
| # | Sequences (R- = HCC(CH2)4CO-) | RT | Purity at 214 nm | MS [M + 1] | ||
|---|---|---|---|---|---|---|
| CH3OH | CH3CN b | m/z (calcd) | m/z (obsd) | |||
| 12 | R-Val(Me)-Val(Me)-Val(Me)-Ala(Me)-Phe(Me)-OH | 6.83 a | 7.19 | >99 | 734.4463 | 734.4468 |
| 15 | R-Val-Val-Val-Ala-Phe-OH | 8.83 a | 7.44 | >99 | 642.3861 | 642.3865 |
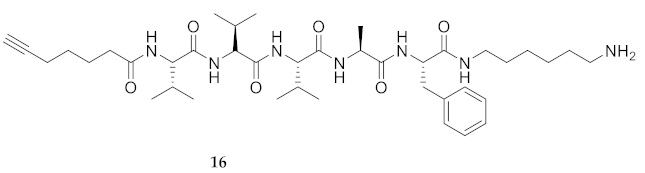
| 16 | R-Val-Val-Val-Ala-Phe-NH(CH2)6NH2 | 7.33 a | 6.52 | >99 | 740.5067 | 740.5069 |
| 17 | R-Val(Me)-Val(Me)-Val(Me)-Ala(Me)-Phe(Me)-NH(CH2)6NH2 | 8.95 a | 7.81 | >99 | 810.5852 | 810.5851 |
| 18 | R-Val(Me)-Val-Val-Ala-Phe-NH(CH2)6NH2 | 8.42 a | 6.90 | >99 | 754.5225 | 754.5232 |
| 19 | R-Val-Val(Me)-Val-Ala-Phe-NH(CH2)6NH2 | 7.86 a | 6.15 | >99 | 754.5225 | 754.5235 |
| 20 | R-Val-Val-Val(Me)-Ala-Phe-NH(CH2)6NH2 | 7.91 a | 6.57 | >99 | 754.5225 | 754.5229 |
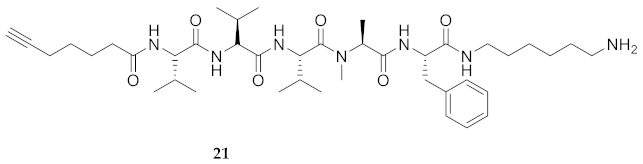
| 21 | R-Val-Val-Val-Ala(Me)-Phe-NH(CH2)6NH2 | 7.61 a | 6.44 | >99 | 754.5225 | 754.5225 |
| 22 | R-Val-Val-Val-Ala-Phe(Me)-NH(CH2)6NH2 | 7.62 a | 6.62 | >99 | 754.5225 | 754.5225 |
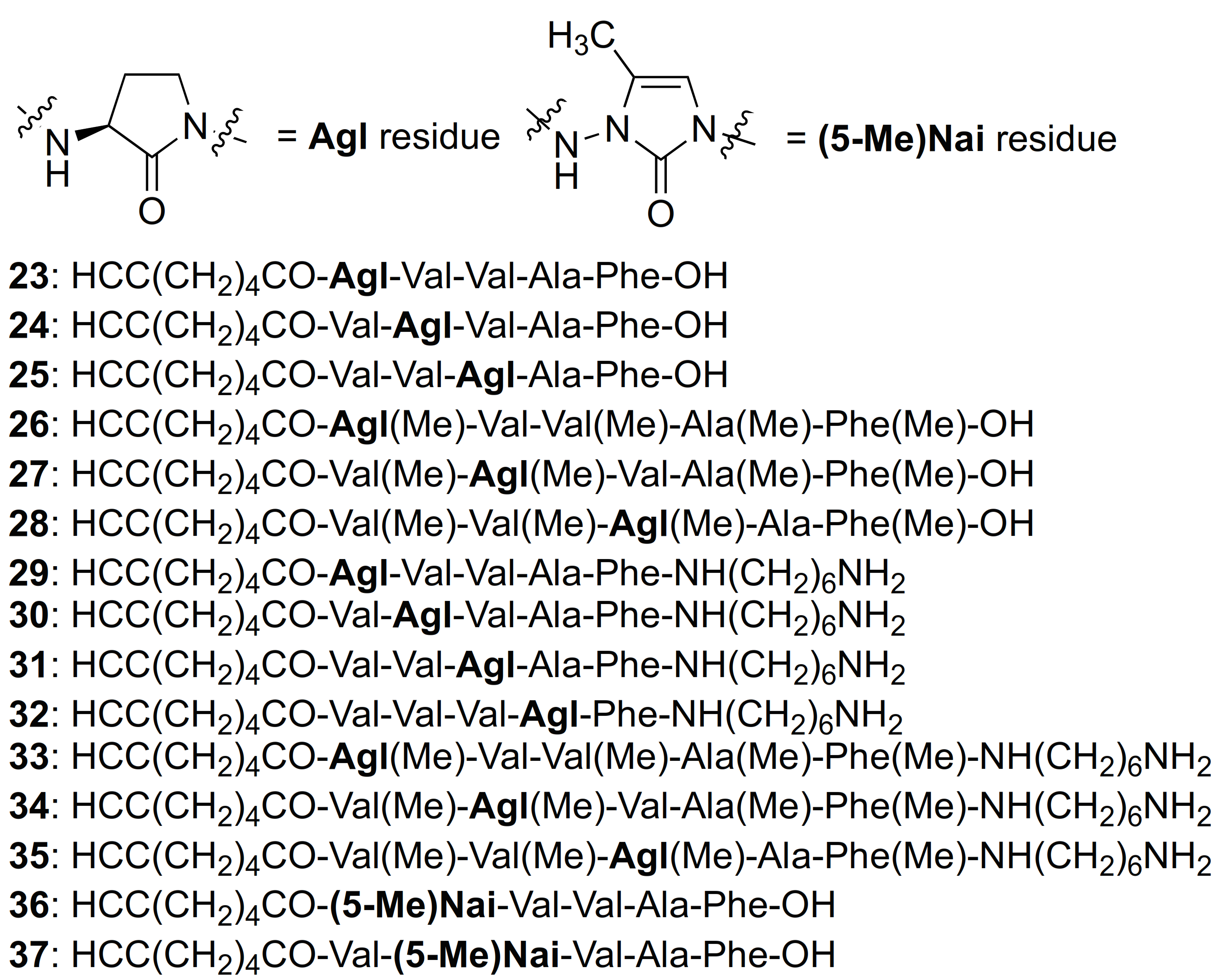
| 23 | R-Agl-Val-Val-Ala-Phe-OH | 7.11 c | 6.71 | >99 | 626.3548 | 626.3523 |
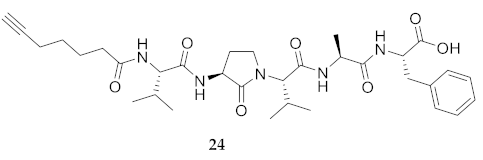
| 24 | R-Val-Agl-Val-Ala-Phe-OH | 7.55 c | 6.87 | >99 | 626.3548 | 626.3529 |
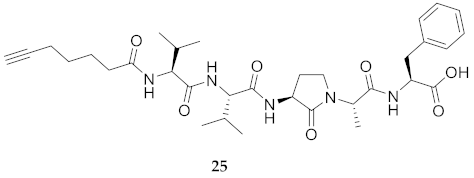
| 25 | R-Val-Val-Agl-Ala-Phe-OH | 7.94 c | 7.17 | >99 | 626.3548 | 626.3521 |
| 26 | R-Agl(Me)-Val-Val(Me)-Ala(Me)-Phe(Me)-OH | 8.69 c | 7.95 | >99 | 682.4174 | 682.4149 |
| 27 | R-Val(Me)-Agl(Me)-Val-Ala(Me)-Phe(Me)-OH | 9.12 c | 8.26 | >99 | 704.3993 d | 704.3999 d |
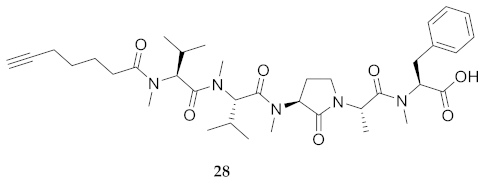
| 28 | R-Val(Me)-Val(Me)-Agl(Me)-Ala-Phe(Me)-OH | 9.01 c | 8.43 | >99 | 704.3993 d | 704.3964 d |
| 29 | R-Agl-Val-Val-Ala-Phe-NH(CH2)6NH2 | 6.72 a | 6.00 | >94 | 724.4756 | 724.4763 |
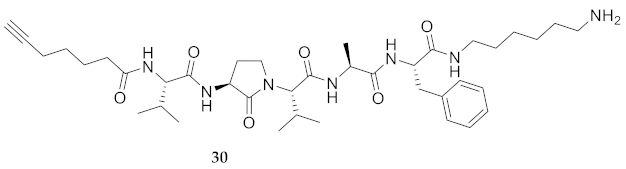
| 30 | R-Val-Agl-Val-Ala-Phe-NH(CH2)6NH2 | 6.83 a | 5.98 | >96 | 724.4756 | 724.4774 |
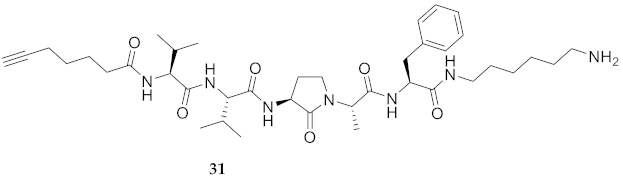
| 31 | R-Val-Val-Agl-Ala-Phe-NH(CH2)6NH2 | 7.50 c | 6.44 | >99 | 724.4756 | 724.4768 |
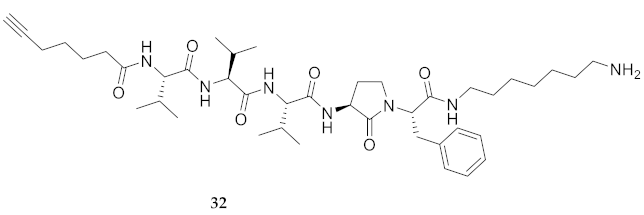
| 32 | R-Val-Val-Val-Agl-Phe-NH(CH2)6NH2 | 5.31 b | 6.42 | >92 | 752.5069 | 752.5082 |
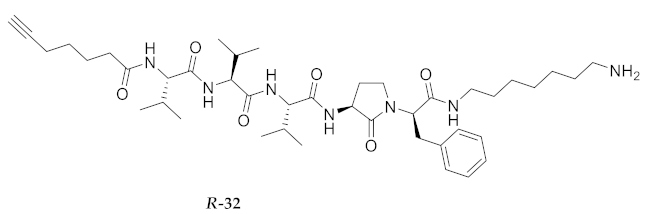
| R-32 | R-Val-Val-Val-Agl-D-Phe-NH(CH2)6NH2 | 5.07 b | 6.42 | >94 | 752.5069 | 752.5072 |
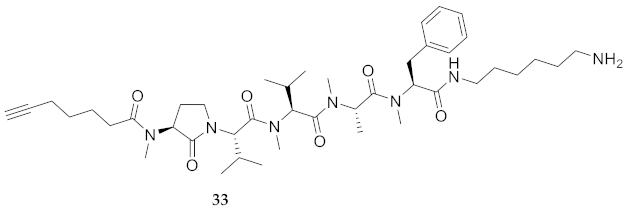
| 33 | R-Agl(Me)-Val-Val(Me)-Ala(Me)-Phe(Me)-NH(CH2)6NH2 | 6.72 c | 7.03 | >99 | 780.5382 | 780.5389 |
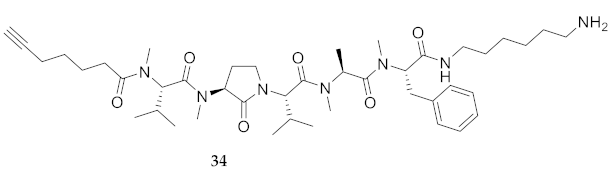
| 34 | R-Val(Me)-Agl(Me)-Val-Ala(Me)-Phe(Me)-NH(CH2)6NH2 | 6.86 c | 7.09 | >99 | 780.5382 | 780.5383 |
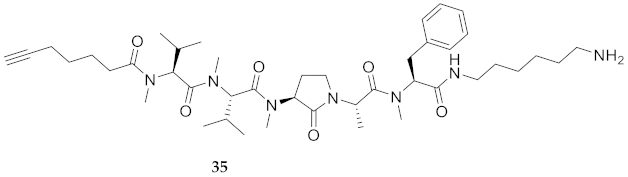
| 35 | R-Val(Me)-Val(Me)-Agl(Me)-Ala-Phe(Me)-NH(CH2)6NH2 | 6.80 c | 7.18 | >99 | 780.5382 | 780.5394 |
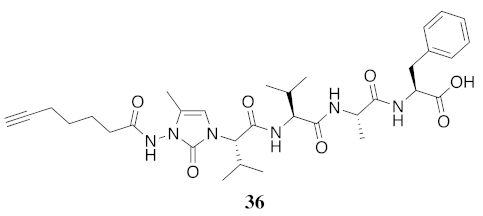
| 36 | R-(5-Me)Nai-Val-Val-Ala-Phe-OH | 5.34 c | 6.48 | >99 | 639.3501 | 639.3474 |
| 37 | R-Val-(5-Me)Nai-Val-Ala-Phe-OH | 4.41 c | 8.68 | >96 | 639.3501 | - |
| # | Sequence (R- = HCC(CH2)4CO-) | Ldi WT 1 (μM) | Ldi Sb-Res 2 (μM) | Ldi MF-Res 3 (μM) | Ldi AmB-Res 4 (μM) | CC50 (μM) | Selectivity Index |
|---|---|---|---|---|---|---|---|
| 12 | R-Val(Me)-Val(Me)-Val(Me)-Ala(Me)-Phe(Me)-OH | 68.33 [61.53, 75.00] | 70.69 [65.62, 75.89] | 75.60 [71.05, 79.15] | 79.59 [73.92, 85.46] | 281 | 4.1 |
| 15 | R-Val-Val-Val-Ala-Phe-OH | 44.69 [34.60, 58.13] | 60.26 [54.09, 66.33] | 50.77 [44.46, 57.93] | 46.38 [41.63, 51.59] | 316 | 7.0 |
| 16 | R-Val-Val-Val-Ala-Phe-NH(CH2)6NH2 | 23.38 [17.49, 32.79] | 30.34 [24.31, 38.73] | 18.09 [16.25, 20.15] | 20.76 [18.22, 23.64] | 446 | 19.0 |
| 17 | R-Val(Me)-Val(Me)-Val(Me)-Ala(Me)-Phe(Me)-NH(CH2)6NH2 | 48.42 [44.44, 52.60] | 60.18 [55.50, 65.25] | 25.46 [21.71, 29.97] | 5.60 [5.06, 6.16] | 338 | 6.9 |
| 18 | R-Val(Me)-Val-Val-Ala-Phe-NH(CH2)6NH2 | 36.70 [33.61, 40.00] | 39.65 [36.65, 42.83] | 12.12 [10.87, 13.58] | 5.03 [4.71, 5.34] | 177 | 4.8 |
| 19 | R-Val-Val(Me)-Val-Ala-Phe-NH(CH2)6NH2 | 81.17 [75.54, 87.30] | 78.91 [74.53, 83.48] | 89.23 [83.11, 95.96] | 70.26 [65.91, 75.02] | 316 | 3.9 |
| 20 | R-Val-Val-Val(Me)-Ala-Phe-NH(CH2)6NH2 | 53.86 [46.63, 62.51] | 40.25 [34.98, 46.35] | 60.64 [55.44, 66.22] | 57.18 [52.23, 62.35] | 281 | 5.2 |
| 21 | R-Val-Val-Val-Ala(Me)-Phe-NH(CH2)6NH2 | 75.83 [70.34, 81.57] | 79.78 [74.41, 85.68] | 63.53 [58.45, 69.01] | 65.66 [59.68, 72.35] | 1440 | 18.9 |
| 22 | R-Val-Val-Val-Ala-Phe(Me)-NH(CH2)6NH2 | 29.04 [24.49, 34.38] | 25.02 [20.45, 30.70] | 27.76 [23.00, 33.13] | 25.91 [21.00, 31.54] | 446 | 15.3 |
| 23 | R-Agl-Val-Val-Ala-Phe-OH | 225.1 [201.5, 261.6] | 240.6 [209.0, 299.3] | 231.5 [203.1, 280.7] | 211.7 [193.1, 238.8] | 363 | 1.61 |
| 24 | R-Val-Agl-Val-Ala-Phe-OH | 248.1 [231.3, 270.5] | 251.1 [233.4, 274.7] | 237.5 [218.6, 263.7] | 261.1 [233.1, 304.7] | 398 | 1.6 |
| 25 | R-Val-Val-Agl-Ala-Phe-OH | N.D. | N.D. | N.D. | N.D. | 416 | - |
| 26 | R-Agl(Me)-Val-Val(Me)-Ala(Me)-Phe(Me)-OH | 217.00 [204.5, 232.8] | 220.4 [209.9, 233.0] | 223.7 [211.1, 239.1] | 252.3 [229.1, 284.9] | 316 | 1.46 |
| 27 | R-Val(Me)-Agl(Me)-Val-Ala(Me)-Phe(Me)-OH | 283.5 [261.8, 314.1] | 295.8 [275.1, 324.4] | N.D. | N.D. | 295 | 1.0 |
| 28 | R-Val(Me)-Val(Me)-Agl(Me)-Ala-Phe(Me)-OH | 254.7 [238.4, 274.5] | 250.2 [232.1, 272.7] | 281.5 [269.8, 295.7] | N.D. | 245 | 0.9 |
| 29 | R-Agl-Val-Val-Ala-Phe-NH(CH2)6NH2 | 74.98 [69.52, 80.41] | 90.39 [86.16, 94.65] | 67.89 [64.58, 71.27] | 71.98 [69.28, 74.69] | 416 | 5.5 |
| 30 | R-Val-Agl-Val-Ala-Phe-NH(CH2)6NH2 | 47.43 [40.21, 55.05] | 37.65 [31.00, 45.19] | 129.3 [125.3, 133.6] | 114.4 [111.4, 117.5] | 407 | 8.6 |
| 31 | R-Val-Val-Agl-Ala-Phe-NH(CH2)6NH2 | 64.64 [59.94, 69.44] | 48.78 [41.26, 56.36] | 126.4 [122.8, 130.1] | 130.20 [128.1, 132.6] | 389 | 6.0 |
| 32 | R-Val-Val-Val-Agl-Phe-NH(CH2)6NH2 | 91.17 [88.95, 93.47] | 87.78 [85.60, 90.02] | 84.21 [82.07, 86.39] | 90.08 [88.22, 91.99] | 389 | 4.3 |
| R-32 | R-Val-Val-Val-Agl-D-Phe-NH(CH2)6NH2 | 92.56 [90.51, 94.66] | 110.8 [106.2, 115.5] | 123.1 [120.5, 125.8] | 117.6 [114.8, 120.4] | 338 | 3.6 |
| 33 | R-Agl(Me)-Val-Val(Me)-Ala(Me)-Phe(Me)-NH(CH2)6NH2 | 119.4 [116.2, 122.8] | 140.4 [138.4, 142.4] | 191.3 [185.1, 198.4] | 183.7 [177.1, 191.1] | 407 | 3.4 |
| 34 | R-Val(Me)-Agl(Me)-Val-Ala(Me)-Phe(Me)-NH(CH2)6NH2 | 33.22 [31.06, 35.49] | 35.75 33.73, 37.88] | 23.13 [20.72, 25.80] | 24.75 [22.12, 27.67] | 398 | 12.0 |
| 35 | R-Val(Me)-Val(Me)-Agl(Me)-Ala-Phe(Me)-NH(CH2)6NH2 | 78.56 [74.91, 82.31] | 81.52 [78.04, 85.14] | 85.30 [80.97, 89.88] | 92.04 [88.01, 96.27] | 371 | 4.7 |
| 36 | R-(5-Me)Nai-Val-Val-Ala-Phe-OH | N.D. | N.D. | N.D. | N.D. | 436 | - |
| 37 | R-Val-(5-Me)Nai-Val-Ala-Phe-OH | N.D. | N.D. | N.D. | N.D. | 407 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.M.T.; Brettell, S.; Douanne, N.; Duquette, C.; Corbeil, A.; Fajardo, E.F.; Olivier, M.; Fernandez-Prada, C.; Lubell, W.D. Influence of N-Methylation and Conformation on Almiramide Anti-Leishmanial Activity. Molecules 2021, 26, 3606. https://doi.org/10.3390/molecules26123606
Nguyen AMT, Brettell S, Douanne N, Duquette C, Corbeil A, Fajardo EF, Olivier M, Fernandez-Prada C, Lubell WD. Influence of N-Methylation and Conformation on Almiramide Anti-Leishmanial Activity. Molecules. 2021; 26(12):3606. https://doi.org/10.3390/molecules26123606
Chicago/Turabian StyleNguyen, Anh Minh Thao, Skye Brettell, Noélie Douanne, Claudia Duquette, Audrey Corbeil, Emanuella F. Fajardo, Martin Olivier, Christopher Fernandez-Prada, and William D. Lubell. 2021. "Influence of N-Methylation and Conformation on Almiramide Anti-Leishmanial Activity" Molecules 26, no. 12: 3606. https://doi.org/10.3390/molecules26123606
APA StyleNguyen, A. M. T., Brettell, S., Douanne, N., Duquette, C., Corbeil, A., Fajardo, E. F., Olivier, M., Fernandez-Prada, C., & Lubell, W. D. (2021). Influence of N-Methylation and Conformation on Almiramide Anti-Leishmanial Activity. Molecules, 26(12), 3606. https://doi.org/10.3390/molecules26123606