
Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer


Abstract
:1. Introduction
2. IGF System Components and Signaling
2.1. IGF-1—Molecular Structure and Regulation
2.2. IGF-1 and Glucose Metabolism
3. IGF-1 and Colorectal Cancer Pathogenesis
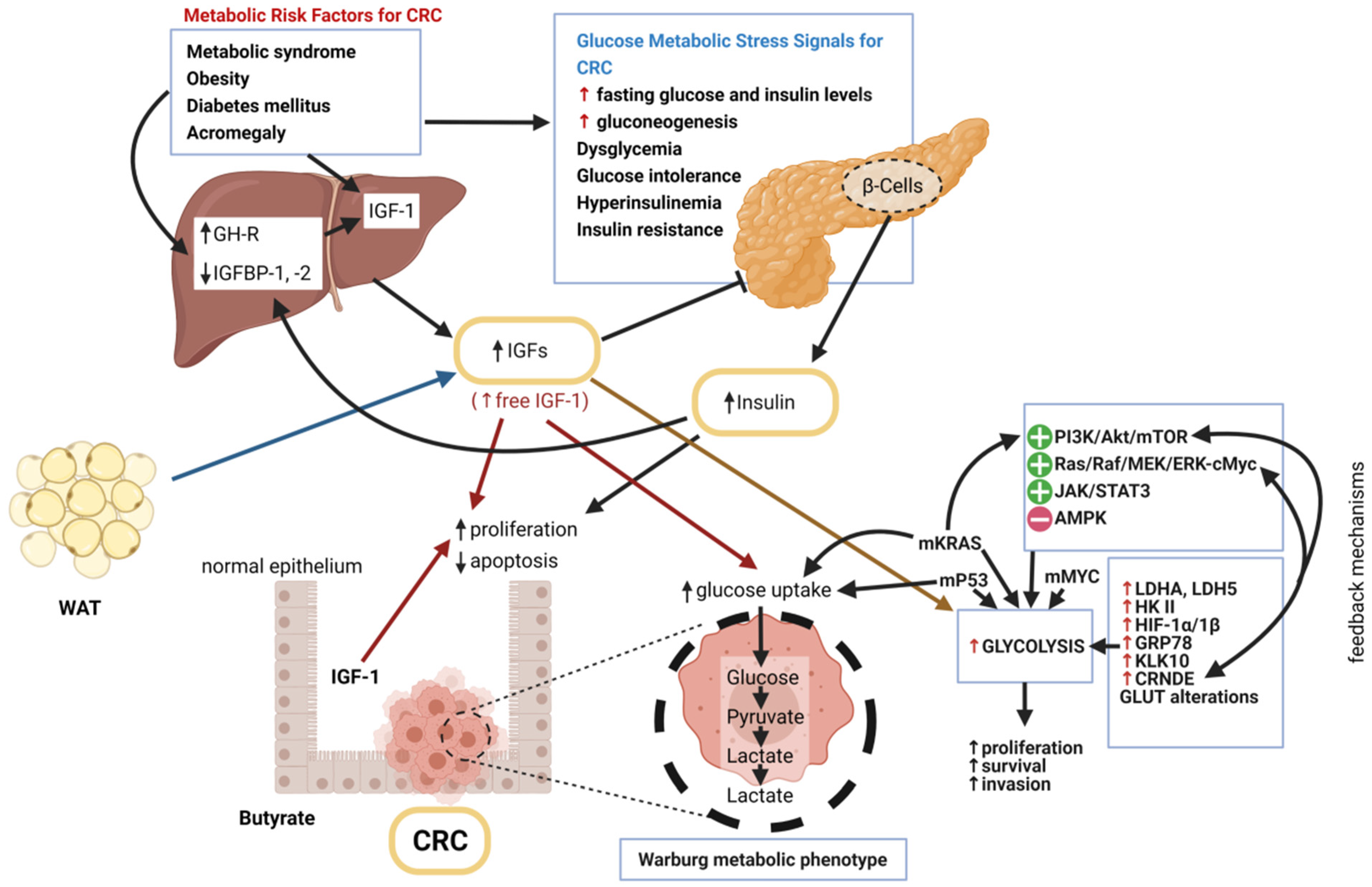
3.1. IGF-1 and Glucose Metabolism Disorders as Risk Factors for Colorectal Cancer
3.1.1. Metabolic Syndrome (MetS)
3.1.2. Obesity
3.1.3. Diabetes Mellitus (DM)
3.1.4. Acromegaly
3.2. IGF-1 and Glucose Metabolism in Normal Colonocytes and CRC Cells
The Role of Insulin/IGF-1 System in Glycolytic Phenotype of CRC Cells
3.3. Genetic Alterations of IGF-1 System Components and Glucose Metabolism in CRC
4. Therapeutic Strategies for Reduction of Metabolic Glucose Disorders in CRC
4.1. Therapeutic Agents Regulating Insulin/IGF Signaling
4.2. Glucose Uptake and Glycolysis Inihbiting Factors
4.2.1. Anti-HIF-1α Factors
4.2.2. Anti-Glucose Transporter Factors (Anti-GLUTs)
4.2.3. Anti-Lactate Dehydrogenase Factors
4.2.4. Anti-Pyruvate-Dehydrogenase (PDH) Complex
4.2.5. Anti-Glucose-Regulated Protein 78 (GRP78)
4.2.6. Other Factors Targeting the Warburg Effect
4.3. Selected Warburg Effect Suppressing Non-Coding RNAs
4.4. Energy Restriction Types and Physical Activity
4.5. Microbiota and the Warburg Effect
5. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-aminoimidazole-4-carboxamide ribonucleotide |
| Akt | serine/threonine-protein kinase or protein kinase B (PKB) |
| AMPK | adenosine monophosphate (AMP)-activated protein kinase |
| APC | adenomatous polyposis coli |
| BMI | body mass index |
| BT | butyrate |
| CaMKK | calcium/calmodulin-dependent proteinase |
| CamKKB | calmodulin kinase kinase B |
| CI | confidence interval |
| cIGF-1 | circulating IGF-1 |
| CIN | chromosome instability |
| CoSCs | colonic stem cells |
| CRC | colorectal cancer |
| CRNDE | colorectal neoplasia differentially expressed gene |
| CSM | cancer-specific mortality |
| DM I, II | diabetes mellitus type 1, type 2 |
| DPP4 | dipeptidyl peptidase 4 |
| ENO1 | enolase 1 |
| FOXO | forkhead box protein |
| ERK | extracellular signal-regulated kinase |
| GA | glutaminase |
| GH | growth hormone |
| GLP-1 | glucagon-like peptide-1 |
| GLUTs | glucose transporters |
| GPI | glycosylphosphatidylinositol |
| GRP78/BiP | G protein-coupled receptor 78/binding immunoglobulin protein |
| HIF-1α | hypoxia-inducible transcription factor 1 alpha |
| HK II | hexokinase II |
| HOMA-IR | homeostatic model assessment-insulin resistance |
| HR | hazard ratio |
| IGF-1, -2 | insulin-like growth factor 1, -2 |
| IGFBPs | IGF binding proteins |
| IGF-1R | IGF receptor type 1 |
| INSR | insulin receptor |
| IR | insulin resistance |
| JAK | Janus kinase |
| KLK10 | kallikrein-related peptidase 10 |
| KRAS | oncogene found in Kirsten rat sarcoma virus |
| LDH5, A, B | lactate dehydrogenase 5, A, B, etc. |
| LKB1 | liver kinase B1 or serine/threonine kinase 11 (STK11) |
| MetS | metabolic syndrome |
| MYC | family of regulator genes/protooncogenes that code for transcription factors |
| NF-κB | nuclear factor-kappa B |
| OD | odds ratio |
| OXPHOS | oxidative phosphorylation |
| PDH | pyruvate dehydrogenase complex |
| PEPCK | phosphoenolpyruvate carboxykinase |
| PFKM | ATP-dependent 6-phosphofructokinase, muscle type |
| PKM2 | pyruvate kinase muscle isozyme 2 |
| PTBP1 | polypyrimidine tract binding protein 1 |
| PTEN | phosphatase and tensin homolog deleted on chromosome ten |
| ROS | reactive oxygen species |
| RR | relative risk |
| SNPs | single nucleotide polymorphisms |
| STAT1/3 | signal transducer and activator of transcription 1/3 |
| STZ | streptozocin |
| TAK1 | TGF-β-activated protein kinase 1 |
| TGF-β | transforming-growth factor beta |
| TMEM219 | the IGFBP-3 receptor |
| TP53 | tumor protein 53 |
| UPC1 | uncoupling protein 1; thermogenin |
| VEGF | vascular endothelial growth factor |
References
- Le Roith, D.; Bondy, C.; Yakar, S.; Liu, J.L.; Butler, A. The somatomedin hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74. [Google Scholar] [CrossRef]
- Jensen-Cody, S.O.; Potthoff, M.J. Hepatokines and metabolism: Deciphering communication from the liver. Mol. Metab. 2020, 44, 101138. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Almengló, C.; Devesa, P. Multiple Effects of Growth Hormone in the Body: Is it Really the Hormone for Growth? Clin. Med. Insights Endocrinol. Diabetes 2016, 9, 47–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemmons, D.R. The relative roles of growth hormone and IGF-1 in controlling insulin sensitivity. J. Clin. Investig. 2004, 113, 25–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Alzhanov, D.; Rotwein, P. Defining human insulin-like growth factor I gene regulation. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E519–E529. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Nava, F.; Lanes, R. GH/IGF-1 Signaling and Current Knowledge of Epigenetics; a Review and Considerations on Possible Therapeutic Options. Int. J. Mol. Sci. 2017, 18, 1624. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.S.; Heald, A.H.; Gibson, J.M.; Cruickshank, J.K.; Dunger, D.B.; Wareham, N.J. Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: A prospective observational study. Lancet 2002, 359, 1740–1745. [Google Scholar] [CrossRef]
- Yakar, S.; Liu, J.L.; Fernandez, A.M.; Wu, Y.; Schally, A.V.; Frystyk, J.; Chernausek, S.D.; Mejia, W.; Le Roith, D. Liver-specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes 2001, 50, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Yakar, S.; Sun, H.; Zhao, H.; Pennisi, P.; Toyoshima, Y.; Setser, J.; Stannard, B.; Scavo, L.; Leroith, D. Metabolic effects of IGF-I deficiency: Lessons from mouse models. Pediatr. Endocrinol. Rev. 2005, 3, 11–19. [Google Scholar]
- Daughaday, W.H. Growth hormone axis overview--somatomedin hypothesis. Pediatr. Nephrol. 2000, 14, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin-IGF axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Sesti, G.; Sciacqua, A.; Cardellini, M.; Marini, M.A.; Maio, R.; Vatrano, M.; Succurro, E.; Lauro, R.; Federici, M.; Perticone, F. Plasma concentration of IGF-I is independently associated with insulin sensitivity in subjects with different degrees of glucose tolerance. Diabetes Care 2005, 28, 120–125. [Google Scholar] [CrossRef] [Green Version]
- De Ita, J.R.; Castilla-Cortázar, I.; Aguirre, G.A.; Sánchez-Yago, C.; Santos-Ruiz, M.O.; Guerra-Menéndez, L.; Martín-Estal, I.; García-Magariño, M.; Lara-Díaz, V.J.; Puche, J.E.; et al. Altered liver expression of genes involved in lipid and glucose metabolism in mice with partial IGF-1 deficiency: An experimental approach to metabolic syndrome. J. Transl. Med. 2015, 13, 326. [Google Scholar] [CrossRef] [Green Version]
- Juul, A.; Scheike, T.; Davidsen, M.; Gyllenborg, J.; Jørgensen, T. Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: A population-based case-control study. Circulation 2002, 106, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Succurro, E.; Andreozzi, F.; Marini, M.A.; Lauro, R.; Hribal, M.L.; Perticone, F.; Sesti, G. Low plasma insulin-like growth factor-1 levels are associated with reduced insulin sensitivity and increased insulin secretion in nondiabetic subjects. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, A.; Cohen, P. Role of insulin-like growth factors and their binding proteins in growth control and carcinogenesis. J. Cell Physiol. 2000, 183, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Khandwala, H.M.; McCutcheon, I.E.; Flyvbjerg, A.; Friend, K.E. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr. Rev. 2000, 21, 215–244. [Google Scholar] [CrossRef]
- Wu, Y.; Yakar, S.; Zhao, L.; Hennighausen, L.; LeRoith, D. Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res. 2002, 62, 1030–1035. [Google Scholar]
- Argon, Y.; Bresson, S.E.; Marzec, M.T.; Grimberg, A. Glucose-Regulated Protein 94 (GRP94): A Novel Regulator of Insulin-Like Growth Factor Production. Cells 2020, 9, 1844. [Google Scholar] [CrossRef]
- Kasprzak, A.; Szaflarski, W. Role of Alternatively Spliced Messenger RNA (mRNA) Isoforms of the Insulin-Like Growth Factor 1 (IGF1) in Selected Human Tumors. Int. J. Mol. Sci. 2020, 21, 6995. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef]
- Muc-Wierzgoń, M.; Nowakowska-Zajdel, E.; Dzięgielewska-Gęsiak, S.; Kokot, T.; Klakla, K.; Fatyga, E.; Grochowska-Niedworok, E.; Waniczek, D.; Wierzgoń, J. Specific metabolic biomarkers as risk and prognostic factors in colorectal cancer. World J. Gastroenterol 2014, 20, 9759–9774. [Google Scholar] [CrossRef]
- Knuppel, A.; Fensom, G.K.; Watts, E.L.; Gunter, M.J.; Murphy, N.; Papier, K.; Perez-Cornago, A.; Schmidt, J.A.; Smith Byrne, K.; Travis, R.C.; et al. Circulating Insulin-like Growth Factor-I Concentrations and Risk of 30 Cancers: Prospective Analyses in UK Biobank. Cancer Res. 2020, 80, 4014–4021. [Google Scholar] [CrossRef]
- Murphy, N.; Carreras-Torres, R.; Song, M.; Chan, A.T.; Martin, R.M.; Papadimitriou, N.; Dimou, N.; Tsilidis, K.K.; Banbury, B.; Bradbury, K.E.; et al. Circulating Levels of Insulin-like Growth Factor 1 and Insulin-like Growth Factor Binding Protein 3 Associate with Risk of Colorectal Cancer Based on Serologic and Mendelian Randomization Analyses. Gastroenterology 2020, 158, 1300–1312.e20. [Google Scholar] [CrossRef] [Green Version]
- Schoen, R.E.; Tangen, C.M.; Kuller, L.H.; Burke, G.L.; Cushman, M.; Tracy, R.P.; Dobs, A.; Savage, P.J. Increased blood glucose and insulin, body size, and incident colorectal cancer. J. Natl. Cancer Inst. 1999, 91, 1147–11454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.W.; Han, D.S.; Park, Y.K.; Son, B.K.; Paik, C.H.; Lee, H.L.; Jeon, Y.C.; Sohn, J.H. Association of obesity, serum glucose and lipids with the risk of advanced colorectal adenoma and cancer: A case-control study in Korea. Dig. Liver Dis. 2006, 38, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Pan, S.; Wen, S.W.; Johnson, K.C. Canadian Cancer Registries Epidemiology Research Group. Physical inactivity, energy intake, obesity and the risk of rectal cancer in Canada. Int. J. Cancer 2003, 105, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Howard, B.V.; Wylie-Rosett, J.; Anderson, G.L.; Ho, G.Y.; et al. Insulin, insulin-like growth factor-I, endogenous estradiol, and risk of colorectal cancer in postmenopausal women. Cancer Res. 2008, 68, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Chiodini, P.; Capuano, A.; Bellastella, G.; Maiorino, M.I.; Rafaniello, C.; Panagiotakos, D.B.; Giugliano, D. Colorectal cancer association with metabolic syndrome and its components: A systematic review with meta-analysis. Endocrine. 2013, 44, 634–647. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Meyerhardt, J.A.; Chan, A.T.; Ng, K.; Chan, J.A.; Wu, K.; Pollak, M.N.; Giovannucci, E.L.; Fuchs, C.S. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 176–185. [Google Scholar] [CrossRef]
- Walkiewicz, K.; Nowakowska-Zajdel, E.; Kozieł, P.; Muc-Wierzgoń, M. The role of some ADAM-proteins and activation of the insulin growth factor-related pathway in colorectal cancer. Cent. Eur. J. Immunol. 2018, 43, 109–113. [Google Scholar] [CrossRef]
- Hu, J.; Liu, X.; Chi, J.; Che, K.; Feng, Y.; Zhao, S.; Wang, Z.; Wang, Y. Expressions of IGF-1, ERK, GLUT4, IRS-1 in metabolic syndrome complicated with colorectal cancer and their associations with the clinical characteristics of CRC. Cancer Biomark. 2018, 21, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Ellis, B.C.; Graham, L.D.; Molloy, P.L. CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism. Biochim. Biophys. Acta 2014, 1843, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Buono, R.; Longo, V.D. Starvation, Stress Resistance, and Cancer. Trends Endocrinol. Metab. 2018, 29, 271–280. [Google Scholar] [CrossRef] [PubMed]
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Liu, X.; Bu, P.; Lin, C.S.; Rakhilin, N.; Locasale, J.W.; Shen, X. A metabolic signature of colon cancer initiating cells. In Proceedings of the 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Chicago, IL, USA, 26–30 August 2014; Volume 2014, pp. 4759–4762. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.E.; Short, S.P.; Williams, C.S. Colorectal Cancer and Metabolism. Curr. Colorectal Cancer Rep. 2018, 14, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef]
- Bathe, O.F.; Farshidfar, F. From genotype to functional phenotype: Unraveling the metabolomic features of colorectal cancer. Genes 2014, 5, 536–560. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Fang, X. Advances in glucose metabolism research in colorectal cancer. Biomed. Rep. 2016, 5, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, O.; Serna-Blasco, R. Targeting KRAS Mutant CMS3 Subtype by Metabolic Inhibitors. Adv. Exp. Med. Biol. 2018, 1110, 23–34. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Humbel, R.E. Insulin-like growth factors I and II. Eur. J. Biochem. 1990, 190, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Ren, H.; Gao, S. Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins: Roles in skeletal muscle growth and differentiation. Gen. Comp. Endocrinol. 2010, 167, 344–351. [Google Scholar] [CrossRef]
- Allard, J.B.; Duan, C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front. Endocrinol. 2018, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Bach, L.A. IGF-binding proteins. J. Mol. Endocrinol. 2018, 61, T11–T28. [Google Scholar] [CrossRef] [Green Version]
- Collett-Solberg, P.F.; Cohen, P. The role of the insulin-like growth factor binding proteins and the IGFBP proteases in modulating IGF action. Endocrinol. Metab. Clin. N. Am. 1996, 25, 591–614. [Google Scholar] [CrossRef]
- Lelbach, A.; Muzes, G.; Feher, J. The insulin-like growth factor system: IGFs, IGF-binding proteins and IGFBP-proteases. Acta Physiol. Hung. 2005, 92, 97–107. [Google Scholar] [CrossRef]
- Clemmons, D.R. Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol. Metab. Clin. N. Am. 2012, 41, 425–443. [Google Scholar] [CrossRef] [Green Version]
- Oberbauer, A.M. The Regulation of IGF-1 Gene Transcription and Splicing during Development and Aging. Front. Endocrinol. 2013, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemmons, D.R. Role of insulin-like growth factor in maintaining normal glucose homeostasis. Horm. Res. 2004, 62, 77–82. [Google Scholar] [CrossRef]
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1447. [Google Scholar] [CrossRef] [PubMed]
- Daughaday, W.H.; Rotwein, P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 1989, 10, 68–91. [Google Scholar] [CrossRef] [PubMed]
- Rinderknecht, E.; Humbel, R.E. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J. Biol. Chem. 1978, 253, 2769–2776. [Google Scholar] [CrossRef]
- Redwan, E.M.; Linjawi, M.H.; Uversky, V.N. Looking at the carcinogenicity of human insulin analogues via the intrinsic disorder prism. Sci. Rep. 2016, 6, 23320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, D.; Simon, M.; Yehezkel, E.; Laron, Z.; Werner, H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab. Res. Rev. 2009, 25, 41–49. [Google Scholar] [CrossRef]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, J.A.M.J.L. New Insights from IGF-IR Stimulating Activity Analyses: Pathological Considerations. Cells 2020, 9, 862. [Google Scholar] [CrossRef] [Green Version]
- Varma Shrivastav, S.; Bhardwaj, A.; Pathak, K.A.; Shrivastav, A. Insulin-Like Growth Factor Binding Protein-3 (IGFBP-3): Unraveling the Role in Mediating IGF-Independent Effects within the Cell. Front. Cell Dev. Biol. 2020, 8, 286. [Google Scholar] [CrossRef]
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef]
- Simpson, H.L.; Jackson, N.C.; Shojaee-Moradie, F.; Jones, R.H.; Russell-Jones, D.L.; Sönksen, P.H.; Dunger, D.B.; Umpleby, A.M. Insulin-like growth factor I has a direct effect on glucose and protein metabolism, but no effect on lipid metabolism in type 1 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Clemmons, D.R. Involvement of insulin-like growth factor-I in the control of glucose homeostasis. Curr. Opin. Pharmacol. 2006, 6, 620–625. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Trefely, S.; Khoo, P.S.; Krycer, J.R.; Chaudhuri, R.; Fazakerley, D.J.; Parker, B.L.; Sultani, G.; Lee, J.; Stephan, J.P.; Torres, E.; et al. Kinome Screen Identifies PFKFB3 and Glucose Metabolism as Important Regulators of the Insulin/Insulin-like Growth Factor (IGF)-1 Signaling Pathway. J. Biol. Chem. 2015, 290, 25834–25846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, E.R.; DeMeo, J.; Lei, H. The insulin-like growth factor (IGF)-I E-peptides are required for isoform-specific gene expression and muscle hypertrophy after local IGF-I production. J. Appl. Physiol. (1985) 2010, 108, 1069–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, B.K.; Barton, E.R. New Modulators for IGF-I Activity within IGF-I Processing Products. Front. Endocrinol. 2013, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Barton, E.R. The ABCs of IGF-I isoforms: Impact on muscle hypertrophy and implications for repair. Appl. Physiol. Nutr. Metab. 2006, 31, 791–797. [Google Scholar] [CrossRef]
- Rotwein, P. Two insulin-like growth factor I messenger RNAs are expressed in human liver. Proc. Natl. Acad. Sci. USA 1986, 83, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Pell, J.M.; Saunders, J.C.; Gilmour, R.S. Differential regulation of transcription initiation from insulin-like growth factor-I (IGF-I) leader exons and of tissue IGF-I expression in response to changed growth hormone and nutritional status in sheep. Endocrinology 1993, 132, 1797–1807. [Google Scholar] [CrossRef]
- Rotwein, P. Mapping the growth hormone--Stat5b--IGF-I transcriptional circuit. Trends Endocrinol. Metab. 2012, 23, 186–193. [Google Scholar] [CrossRef] [Green Version]
- D’Ercole, A.J.; Stiles, A.D.; Underwood, L.E. Tissue concentrations of somatomedin C: Further evidence for multiple sites of synthesis and paracrine or autocrine mechanisms of action. Proc. Natl. Acad. Sci. USA 1984, 81, 935–939. [Google Scholar] [CrossRef] [Green Version]
- Bach, M.A.; Shen-Orr, Z.; Lowe, W.L., Jr.; Roberts, C.T., Jr.; LeRoith, D. Insulin-like growth factor I mRNA levels are developmentally regulated in specific regions of the rat brain. Brain Res. Mol. Brain Res. 1991, 10, 43–48. [Google Scholar] [CrossRef]
- Lobie, P.E.; Zhu, T.; Graichen, R.; Goh, E.L. Growth hormone, insulin-like growth factor I and the CNS: Localization, function and mechanism of action. Growth Horm. IGF Res. 2000, 10 (Suppl. B), S51–S56. [Google Scholar] [CrossRef]
- Kooijman, R.; Willems, M.; De Haas, C.J.; Rijkers, G.T.; Schuurmans, A.L.; Van Buul-Offers, S.C.; Heijnen, C.J.; Zegers, B.J. Expression of type I insulin-like growth factor receptors on human peripheral blood mononuclear cells. Endocrinology 1992, 131, 2244–2250. [Google Scholar] [CrossRef] [Green Version]
- Weigent, D.A.; Baxter, J.B.; Blalock, J.E. The production of growth hormone and insulin-like growth factor-I by the same subpopulation of rat mononuclear leukocytes. Brain Behav. Immun. 1992, 6, 365–376. [Google Scholar] [CrossRef]
- Fragala, M.S.; Jajtner, A.R.; Townsend, J.R.; Gonzalez, A.M.; Wells, A.J.; Oliveira, L.P.; Hoffman, J.R.; Stout, J.R.; Fukuda, D.H. Leukocyte IGF-1 receptor expression during muscle recovery. Med. Sci. Sports Exerc. 2015, 47, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilakos, G.; Lei, H.; Yang, Y.; Puglise, J.; Matheny, M.; Durzynska, J.; Ozery, M.; Bennett, K.; Spradlin, R.; Bonanno, H.; et al. Deletion of muscle IGF-I transiently impairs growth and progressively disrupts glucose homeostasis in male mice. FASEB J. 2019, 33, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustina, A.; Berardelli, R.; Gazzaruso, C.; Mazziotti, G. Insulin and GH-IGF-I axis: Endocrine pacer or endocrine disruptor? Acta Diabetol. 2015, 52, 433–443. [Google Scholar] [CrossRef]
- Bikle, D.D.; Tahimic, C.; Chang, W.; Wang, Y.; Philippou, A.; Barton, E.R. Role of IGF-I signaling in muscle bone interactions. Bone 2015, 80, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Goya, L.; de la Puente, A.; Ramos, S.; Martín, M.A.; Escrivá, F.; Pascual-Leone, A.M. Regulation of insulin-like growth factor-I and -II by glucose in primary cultures of fetal rat hepatocytes. J. Biol. Chem. 1999, 274, 24633–24640. [Google Scholar] [CrossRef] [Green Version]
- Goya, L.; de la Puente, A.; Ramos, S.; Martín, M.A.; Escrivá, F.; Alvarez, C.; Pascual-Leone, A.M. Regulation of IGF-I and -II by insulin in primary cultures of fetal rat hepatocytes. Endocrinology 2001, 142, 5089–5096. [Google Scholar] [CrossRef]
- Dogansen, S.C.; Yalin, G.Y.; Tanrikulu, S.; Yarman, S. Impact of Glucose Metabolism Disorders on IGF-1 Levels in Patients with Acromegaly. Horm. Metab. Res. 2018, 50, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, Y.; Handa, H.; Imai, T. Arginine induces IGF-1 secretion from the endoplasmic reticulum. Biochem. Biophys. Res. Commun. 2019, 514, 1128–1132. [Google Scholar] [CrossRef]
- Kanbur-Oksüz, N.; Derman, O.; Kinik, E. Correlation of sex steroids with IGF-1 and IGFBP-3 during different pubertal stages. Turk. J. Pediatr. 2004, 46, 315–321. [Google Scholar] [PubMed]
- Zhang, W.B.; Aleksic, S.; Gao, T.; Weiss, E.F.; Demetriou, E.; Verghese, J.; Holtzer, R.; Barzilai, N.; Milman, S. Insulin-like Growth Factor-1 and IGF Binding Proteins Predict All-Cause Mortality and Morbidity in Older Adults. Cells 2020, 9, 1368. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C. Insulin-like growth factor-I (IGF-I) and clinical nutrition. Clin. Sci. 2013, 125, 265–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, A.; Gupta, R.; Patel, S.; Velingkaar, N.; Kondratov, R. Cryptochromes regulate IGF-1 production and signaling through control of JAK2-dependent STAT5B phosphorylation. Mol. Biol. Cell 2017, 28, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Charles, J.F. Gut Microbiota and IGF-1. Calcif. Tissue Int. 2018, 102, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Vasques, G.A.; Andrade, N.L.M.; Correa, F.A.; Jorge, A.A.L. Update on new GH-IGF axis genetic defects. Arch. Endocrinol. Metab. 2019, 63, 608–617. [Google Scholar] [CrossRef]
- Adamek, A.; Kasprzak, A.; Mikoś, H.; Przybyszewska, W.; Seraszek-Jaros, A.; Czajka, A.; Sterzyńska, K.; Mozer-Lisewska, I. The insulin-like growth factor-1 and expression of its binding protein-3 in chronic hepatitis C and hepatocellular carcinoma. Oncol. Rep. 2013, 30, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- El-Ashmawy, N.E.; El-Bahrawy, H.A.; Shamloula, M.M.; El-Feky, O.A. Biochemical/metabolic changes associated with hepatocellular carcinoma development in mice. Tumour Biol. 2014, 35, 5459–5466. [Google Scholar] [CrossRef] [PubMed]
- Carotti, S.; Guarino, M.P.L.; Valentini, F.; Porzio, S.; Vespasiani-Gentilucci, U.; Perrone, G.; Zingariello, M.; Gallo, P.; Cicala, M.; Picardi, A.; et al. Impairment of GH/IGF-1 Axis in the Liver of Patients with HCV-Related Chronic Hepatitis. Horm. Metab. Res. 2018, 50, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Juul, A. Serum levels of insulin-like growth factor I and its binding proteins in health and disease. Growth Horm. IGF Res. 2003, 13, 113–170. [Google Scholar] [CrossRef]
- Bianda, T.L.; Hussain, M.A.; Keller, A.; Glatz, Y.; Schmitz, O.; Christiansen, J.S.; Alberti, K.G.; Froesch, E.R. Insulin-like growth factor-I in man enhances lipid mobilization and oxidation induced by a growth hormone pulse. Diabetologia 1996, 39, 961–969. [Google Scholar] [CrossRef]
- Russell-Jones, D.L.; Bates, A.T.; Umpleby, A.M.; Hennessy, T.R.; Bowes, S.B.; Hopkins, K.D.; Jackson, N.; Kelly, J.; Shojaee-Moradie, F.; Jones, R.H.; et al. A comparison of the effects of IGF-I and insulin on glucose metabolism, fat metabolism and the cardiovascular system in normal human volunteers. Eur. J. Clin. Investig. 1995, 25, 403–411. [Google Scholar] [CrossRef]
- Böni-Schnetzler, M.; Hauri, C.; Zapf, J. Leptin is suppressed during infusion of recombinant human insulin-like growth factor I (rhIGF I) in normal rats. Diabetologia 1999, 42, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Guler, H.P.; Zapf, J.; Froesch, E.R. Short-term metabolic effects of recombinant human insulin-like growth factor I in healthy adults. N. Engl. J. Med. 1987, 317, 137–140. [Google Scholar] [CrossRef]
- Holt, R.I.; Simpson, H.L.; Sönksen, P.H. The role of the growth hormone-insulin-like growth factor axis in glucose homeostasis. Diabet. Med. 2003, 20, 3–15. [Google Scholar] [CrossRef]
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor 1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Laager, R.; Ninnis, R.; Keller, U. Comparison of the effects of recombinant human insulin-like growth factor-I and insulin on glucose and leucine kinetics in humans. J. Clin. Investig. 1993, 92, 1903–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenobi, P.D.; Graf, S.; Ursprung, H.; Froesch, E.R. Effects of insulin-like growth factor-I on glucose tolerance, insulin levels, and insulin secretion. J. Clin. Investig. 1992, 89, 1908–1913. [Google Scholar] [CrossRef] [PubMed]
- Saukkonen, T.; Shojaee-Moradie, F.; Williams, R.M.; Amin, R.; Yuen, K.C.; Watts, A.; Acerini, C.L.; Umpleby, A.M.; Dunger, D.B. Effects of recombinant human IGF-I/IGF-binding protein-3 complex on glucose and glycerol metabolism in type 1 diabetes. Diabetes 2006, 55, 2365–2370. [Google Scholar] [CrossRef] [Green Version]
- Moses, A.C.; Young, S.C.; Morrow, L.A.; O’Brien, M.; Clemmons, D.R. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes 1996, 45, 91–100. [Google Scholar] [CrossRef]
- Sjögren, K.; Wallenius, K.; Liu, J.L.; Bohlooly-Y, M.; Pacini, G.; Svensson, L.; Törnell, J.; Isaksson, O.G.; Ahrén, B.; Jansson, J.O.; et al. Liver-derived IGF-I is of importance for normal carbohydrate and lipid metabolism. Diabetes 2001, 50, 1539–1545. [Google Scholar] [CrossRef] [Green Version]
- Crosby, P.; Hamnett, R.; Putker, M.; Hoyle, N.P.; Reed, M.; Karam, C.J.; Maywood, E.S.; Stangherlin, A.; Chesham, J.E.; Hayter, E.A.; et al. Insulin/IGF-1 Drives PERIOD Synthesis to Entrain Circadian Rhythms with Feeding Time. Cell 2019, 177, 896–909.e20. [Google Scholar] [CrossRef] [Green Version]
- Isley, W.L.; Underwood, L.E.; Clemmons, D.R. Dietary components that regulate serum somatomedin-C concentrations in humans. J. Clin. Investig. 1983, 71, 175–182. [Google Scholar] [CrossRef]
- Aguirre, G.A.; De Ita, J.R.; de la Garza, R.G.; Castilla-Cortazar, I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J. Transl. Med. 2016, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Kasprzak, A.; Kwasniewski, W.; Adamek, A.; Gozdzicka-Jozefiak, A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat. Res. Rev. Mutat. Res. 2017, 772, 78–104. [Google Scholar] [CrossRef]
- Ma, J.; Pollak, M.N.; Giovannucci, E.; Chan, J.M.; Tao, Y.; Hennekens, C.H.; Stampfer, M.J. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J. Natl. Cancer Inst. 1999, 91, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Katki, H.; Graubard, B.; Pollak, M.; Martin, M.; Tao, Y.; Schoen, R.E.; Church, T.; Hayes, R.B.; Greene, M.H.; et al. Serum IGF1, IGF2 and IGFBP3 and risk of advanced colorectal adenoma. Int. J. Cancer 2012, 131, E105–E113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Zhang, X.; Du, L.L.; Wang, Y.; Liu, D.B.; Han, C.Z.; Jing, J.X.; Zhao, X.W.; Xu, X.Q. Possible roles of insulin, IGF-1 and IGFBPs in initiation and progression of colorectal cancer. World J. Gastroenterol. 2014, 20, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Pollak, M.N.; Platz, E.A.; Willett, W.C.; Stampfer, M.J.; Majeed, N.; Colditz, G.A.; Speizer, F.E.; Hankinson, S.E. A prospective study of plasma insulin-like growth factor-1 and binding protein-3 and risk of colorectal neoplasia in women. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 345–349. [Google Scholar] [PubMed]
- Kushlinskii, N.E.; Gershtein, E.S.; Nikolaev, A.A.; Delektorskaya, V.V.; Korotkova, E.A.; Dvorova, E.K.; Kostyleva, O.I. Insulin-like growth factors (IGF), IGF-binding proteins (IGFBP), and vascular endothelial growth factor (VEGF) in blood serum of patients with colorectal cancer. Bull. Exp. Biol. Med. 2014, 156, 684–688. [Google Scholar] [CrossRef]
- Rinaldi, S.; Cleveland, R.; Norat, T.; Biessy, C.; Rohrmann, S.; Linseisen, J.; Boeing, H.; Pischon, T.; Panico, S.; Agnoli, C.; et al. Serum levels of IGF-I, IGFBP-3 and colorectal cancer risk: Results from the EPIC cohort, plus a meta-analysis of prospective studies. Int. J. Cancer 2010, 126, 1702–1715. [Google Scholar] [CrossRef]
- Kaaks, R.; Toniolo, P.; Akhmedkhanov, A.; Lukanova, A.; Biessy, C.; Dechaud, H.; Rinaldi, S.; Zeleniuch-Jacquotte, A.; Shore, R.E.; Riboli, E. Serum C-peptide, insulin-like growth factor (IGF)-I, IGF-binding proteins, and colorectal cancer risk in women. J. Natl. Cancer Inst. 2000, 92, 1592–1600. [Google Scholar] [CrossRef]
- Sandhu, M.S.; Dunger, D.B.; Giovannucci, E.L. Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J. Natl. Cancer Inst. 2002, 94, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Peters, G.; Gongoll, S.; Langner, C.; Mengel, M.; Piso, P.; Klempnauer, J.; Rüschoff, J.; Kreipe, H.; von Wasielewski, R. IGF-1R, IGF-1 and IGF-2 expression as potential prognostic and predictive markers in colorectal-cancer. Virchows Arch. 2003, 443, 139–145. [Google Scholar] [CrossRef]
- Kasprzak, A.; Szaflarski, W.; Szmeja, J.; Andrzejewska, M.; Przybyszewska, W.; Kaczmarek, E.; Koczorowska, M.; Kościński, T.; Zabel, M.; Drews, M. Differential expression of IGF-1 mRNA isoforms in colorectal carcinoma and normal colon tissue. Int. J. Oncol. 2013, 42, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Shiratsuchi, I.; Akagi, Y.; Kawahara, A.; Kinugasa, T.; Romeo, K.; Yoshida, T.; Ryu, Y.; Gotanda, Y.; Kage, M.; Shirouzu, K. Expression of IGF-1 and IGF-1R and their relation to clinicopathological factors in colorectal cancer. Anticancer Res. 2011, 31, 2541–2545. [Google Scholar]
- Nosho, K.; Yamamoto, H.; Taniguchi, H.; Adachi, Y.; Yoshida, Y.; Arimura, Y.; Endo, T.; Hinoda, Y.; Imai, K. Interplay of insulin-like growth factor-II, insulin-like growth factor-I, insulin-like growth factor-I receptor, COX-2, and matrix metalloproteinase-7, play key roles in the early stage of colorectal carcinogenesis. Clin. Cancer Res. 2004, 10, 7950–7957. [Google Scholar] [CrossRef] [Green Version]
- Michell, N.P.; Langman, M.J.; Eggo, M.C. Insulin-like growth factors and their binding proteins in human colonocytes: Preferential degradation of insulin-like growth factor binding protein 2 in colonic cancers. Br. J. Cancer 1997, 76, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Freier, S.; Weiss, O.; Eran, M.; Flyvbjerg, A.; Dahan, R.; Nephesh, I.; Safra, T.; Shiloni, E.; Raz, I. Expression of the insulin-like growth factors and their receptors in adenocarcinoma of the colon. Gut 1999, 44, 704–708. [Google Scholar] [CrossRef]
- Yamamoto, N.; Oshima, T.; Yoshihara, K.; Aoyama, T.; Hayashi, T.; Yamada, T.; Sato, T.; Shiozawa, M.; Yoshikawa, T.; Morinaga, S.; et al. Clinicopathological significance and impact on outcomes of the gene expression levels of IGF-1, IGF-2 and IGF-1R, IGFBP-3 in patients with colorectal cancer: Overexpression of the IGFBP-3 gene is an effective predictor of outcomes in patients with colorectal cancer. Oncol. Lett. 2017, 13, 3958–3966. [Google Scholar] [CrossRef]
- Watkins, L.F.; Lewis, L.R.; Levine, A.E. Characterization of the synergistic effect of insulin and transferrin and the regulation of their receptors on a human colon carcinoma cell line. Int. J. Cancer 1990, 45, 372–375. [Google Scholar] [CrossRef]
- Guo, Y.S.; Narayan, S.; Yallampalli, C.; Singh, P. Characterization of insulinlike growth factor I receptors in human colon cancer. Gastroenterology 1992, 102, 1101–1108. [Google Scholar] [CrossRef]
- Giovannucci, E. Insulin, insulin-like growth factors and colon cancer: A review of the evidence. J. Nutr. 2001, 131, 3109S–3120S. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Y.; Wang, L.; Song, Z.Y.; Qu, X.J. Knockdown of type I insulin-like growth factor receptor inhibits human colorectal cancer cell growth and downstream PI3K/Akt, WNT/β-catenin signal pathways. Biomed. Pharmacother. 2015, 73, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Heckl, S.M.; Pellinghaus, M.; Krüger, S.; Bosselmann, C.; Wilhelm, F.; Behrens, H.M.; Schreiber, S.; Röcken, C. Epithelial insulin receptor expression-prognostic relevance in colorectal cancer. Oncotarget 2018, 9, 37497–37508. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z.; Werner, H. Insulin: A Growth Hormone and Potential Oncogene. Pediatr. Endocrinol. Rev. 2020, 17 (Suppl. 1), 191–197. [Google Scholar] [CrossRef]
- Lahm, H.; Suardet, L.; Laurent, P.L.; Fischer, J.R.; Ceyhan, A.; Givel, J.C.; Odartchenko, N. Growth regulation and co-stimulation of human colorectal cancer cell lines by insulin-like growth factor I, II and transforming growth factor alpha. Br. J. Cancer 1992, 65, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lillich, F.F.; Imig, J.D.; Proschak, E. Multi-Target Approaches in Metabolic Syndrome. Front. Pharmacol. 2021, 11, 554961. [Google Scholar] [CrossRef]
- Crawley, D.J.; Holmberg, L.; Melvin, J.C.; Loda, M.; Chowdhury, S.; Rudman, S.M.; Van Hemelrijck, M. Serum glucose and risk of cancer: A meta-analysis. BMC Cancer 2014, 14, 985. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Araki, S.; Tamura, M.; Sakai, I.; Takahashi, Y.; Kashihara, H.; Kono, S. Relation of serum total cholesterol, serum triglycerides and fasting plasma glucose to colorectal carcinoma in situ. Int. J. Epidemiol. 1998, 27, 794–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limburg, P.J.; Stolzenberg-Solomon, R.Z.; Vierkant, R.A.; Roberts, K.; Sellers, T.A.; Taylor, P.R.; Virtamo, J.; Cerhan, J.R.; Albanes, D. Insulin, glucose, insulin resistance, and incident colorectal cancer in male smokers. Clin. Gastroenterol. Hepatol. 2006, 4, 1514–1521. [Google Scholar] [CrossRef] [Green Version]
- Kabat, G.C.; Kim, M.Y.; Strickler, H.D.; Shikany, J.M.; Lane, D.; Luo, J.; Ning, Y.; Gunter, M.J.; Rohan, T.E. A longitudinal study of serum insulin and glucose levels in relation to colorectal cancer risk among postmenopausal women. Br. J. Cancer 2012, 106, 227–232. [Google Scholar] [CrossRef]
- Vulcan, A.; Manjer, J.; Ohlsson, B. High blood glucose levels are associated with higher risk of colon cancer in men: A cohort study. BMC Cancer 2017, 17, 842. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ye, Y.; Wu, H.; Duerksen-Hughes, P.; Zhang, H.; Li, P.; Huang, J.; Yang, J.; Wu, Y.; Xia, D. Association between markers of glucose metabolism and risk of colorectal cancer. BMJ Open 2016, 6, e011430. [Google Scholar] [CrossRef] [Green Version]
- Frystyk, J.; Vestbo, E.; Skjaerbaek, C.; Mogensen, C.E.; Orskov, H. Free insulin-like growth factors in human obesity. Metabolism 1995, 44, 37–44. [Google Scholar] [CrossRef]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisani, P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch. Physiol. Biochem. 2008, 114, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol. Rev. 2015, 95, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Pallavi, R.; Giorgio, M.; Pelicci, P.G. Insights into the beneficial effect of caloric/ dietary restriction for a healthy and prolonged life. Front. Physiol. 2012, 3, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.; Cui, Z.Z.; Zhu, L.; Fu, S.P.; Rossi, M.; Cui, Y.H.; Zhu, B.M. Central IGF1 improves glucose tolerance and insulin sensitivity in mice. Nutr. Diabetes 2017, 7, 2, Erratum in Nutr. Diabetes 2021, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Fenton, J.I.; Birmingham, J.M. Adipokine regulation of colon cancer: Adiponectin attenuates interleukin-6-induced colon carcinoma cell proliferation via STAT-3. Mol. Carcinog. 2010, 49, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Clauson, P.G.; Brismar, K.; Hall, K.; Linnarsson, R.; Grill, V. Insulin-like growth factor-I and insulin-like growth factor binding protein-1 in a representative population of type 2 diabetic patients in Sweden. Scand. J. Clin. Lab. Investig. 1998, 58, 353–360. [Google Scholar] [CrossRef]
- Park, H.; Cho, S.; Woo, H.; Park, S.K.; Shin, H.R.; Chang, S.H.; Yoo, K.Y.; Shin, A. Fasting glucose and risk of colorectal cancer in the Korean Multi-center Cancer Cohort. PLoS ONE 2017, 12, e0188465. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Kartsonaki, C.; Guo, Y.; Chen, Y.; Yang, L.; Bian, Z.; Bragg, F.; Millwood, I.Y.; Shen, L.; Zhou, S.; et al. Diabetes, plasma glucose and incidence of colorectal cancer in Chinese adults: A prospective study of 0.5 million people. J. Epidemiol. Community Health 2018, 72, 919–925. [Google Scholar] [CrossRef] [Green Version]
- Suh, S.; Kim, K.W. Diabetes and Cancer: Cancer Should Be Screened in Routine Diabetes Assessment. Diabetes Metab. J. 2019, 43, 733–743. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928, Erratum in Nat. Rev. Cancer 2009, 9, 224. [Google Scholar] [CrossRef]
- Cao, H.; Jin, C.; Huang, D.; Liu, C.; Sun, D.; Tan, C.; Zhu, X.; Fei, Y. Changes in serum IGF-1 level and tumor VEGF expression in mice with colorectal cancer under hyperglycemic conditions. Mol. Med. Rep. 2013, 7, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.X.; Zhu, H.H.; Zhu, Y.M. Diabetes and cancer: Associations, mechanisms, and implications for medical practice. World J. Diabetes 2014, 5, 372–380. [Google Scholar] [CrossRef]
- Luo, J.; Lin, H.C.; He, K.; Hendryx, M. Diabetes and prognosis in older persons with colorectal cancer. Br. J. Cancer 2014, 110, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Peeters, P.J.; Bazelier, M.T.; Leufkens, H.G.; de Vries, F.; De Bruin, M.L. The risk of colorectal cancer in patients with type 2 diabetes: Associations with treatment stage and obesity. Diabetes Care 2015, 38, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Li, C.; Tang, J.; Yi, C.; Liu, J.Y.; Qiu, M. Higher Expression of Proteins in IGF/IR Axes in Colorectal Cancer is Associated with Type 2 Diabetes Mellitus. Pathol. Oncol. Res. 2016, 22, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ben, Q.; Shen, H.; Lu, W.; Zhang, Y.; Zhu, J. Diabetes mellitus and incidence and mortality of colorectal cancer: A systematic review and meta-analysis of cohort studies. Eur. J. Epidemiol. 2011, 26, 863–876. [Google Scholar] [CrossRef]
- Mills, K.T.; Bellows, C.F.; Hoffman, A.E.; Kelly, T.N.; Gagliardi, G. Diabetes mellitus and colorectal cancer prognosis: A meta-analysis. Dis. Colon Rectum 2013, 56, 1304–1319. [Google Scholar] [CrossRef] [Green Version]
- Petrelli, F.; Ghidini, M.; Rausa, E.; Ghidini, A.; Cabiddu, M.; Borgonovo, K.; Ghilardi, M.; Parati, M.C.; Pietrantonio, F.; Sganzerla, P.; et al. Survival of Colorectal Cancer Patients with Diabetes Mellitus: A Meta-Analysis. Can. J. Diabetes 2021, 45, 186–197.e2. [Google Scholar] [CrossRef]
- Han, X.; Hou, S.; Yang, A. Correlation Between IGFs-Related Proteins Expression and Incidence of Colorectal Cancer in Diabetic Patients and Related Mechanisms. Med. Sci. Monit. 2016, 22, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Hu, L.L.; Sun, A.; Cao, Y.J.; Tang, T.; Zhang, X.P.; Zhang, Q.H. mRNA expression of IGF-1 and IGF-1R in patients with colorectal adenocarcinoma and type 2 diabetes. Arch. Med. Res. 2014, 45, 318–324. [Google Scholar] [CrossRef] [PubMed]
- D’Addio, F.; La Rosa, S.; Maestroni, A.; Jung, P.; Orsenigo, E.; Ben Nasr, M.; Tezza, S.; Bassi, R.; Finzi, G.; Marando, A.; et al. Circulating IGF-I and IGFBP3 Levels Control Human Colonic Stem Cell Function and Are Disrupted in Diabetic Enteropathy. Cell Stem Cell 2015, 17, 486–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, K.; Kubota, M.; Shimizu, M.; Moriwaki, H.; Kuno, T.; Tanaka, T.; Hara, A.; Hirose, Y. C57BL/KsJ-db/db-Apc mice exhibit an increased incidence of intestinal neoplasms. Int. J. Mol. Sci. 2011, 12, 8133–8145. [Google Scholar] [CrossRef] [PubMed]
- Rachdaoui, N. Insulin: The Friend and the Foe in the Development of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 1770. [Google Scholar] [CrossRef] [Green Version]
- Fagin, J.A.; Roberts, C.T., Jr.; LeRoith, D.; Brown, A.T. Coordinate decrease of tissue insulinlike growth factor I posttranscriptional alternative mRNA transcripts in diabetes mellitus. Diabetes 1989, 38, 428–434. [Google Scholar] [CrossRef]
- Zhao, A.Z.; Zhao, H.; Teague, J.; Fujimoto, W.; Beavo, J.A. Attenuation of insulin secretion by insulin-like growth factor 1 is mediated through activation of phosphodiesterase 3B. Proc. Natl. Acad. Sci. USA 1997, 94, 3223–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guler, H.P.; Schmid, C.; Zapf, J.; Froesch, E.R. Effects of recombinant insulin-like growth factor I on insulin secretion and renal function in normal human subjects. Proc. Natl. Acad. Sci. USA 1989, 86, 2868–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, A.; Tovar, A.P.; Castellanos, R.; Valenzuela, A.; Giraldo, C.M.; Pinedo, A.C.; Guerrero, D.P.; Barrera, C.A.; Franco, H.I.; Ribeiro-Oliveira, A., Jr.; et al. Challenges in the diagnosis and management of acromegaly: A focus on comorbidities. Pituitary 2016, 19, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Colao, A.; Grasso, L.F.S.; Giustina, A.; Melmed, S.; Chanson, P.; Pereira, A.M.; Pivonello, R. Acromegaly. Nat. Rev. Dis. Primers 2019, 5, 20, Erratum in Nat. Rev. Dis. Primers 2019, 5, 72. [Google Scholar] [CrossRef]
- Cheng, S.; Al-Agha, R.; Araujo, P.B.; Serri, O.; L Asa, S.; Ezzat, S. Metabolic glucose status and pituitary pathology portend therapeutic outcomes in acromegaly. PLoS ONE 2013, 8, e73543. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, D.; Purice, M.; Coculescu, M. Insulin-like growth factor-I correlates more closely than growth hormone with insulin resistance and glucose intolerance in patients with acromegaly. Pituitary 2013, 16, 168–174. [Google Scholar] [CrossRef]
- Maione, L.; Brue, T.; Beckers, A.; Delemer, B.; Petrossians, P.; Borson-Chazot, F.; Chabre, O.; François, P.; Bertherat, J.; Cortet-Rudelli, C.; et al. French Acromegaly Registry Group. Changes in the management and comorbidities of acromegaly over three decades: The French Acromegaly Registry. Eur. J. Endocrinol. 2017, 176, 645–655. [Google Scholar] [CrossRef]
- Hannon, A.M.; Thompson, C.J.; Sherlock, M. Diabetes in Patients with Acromegaly. Curr. Diab. Rep. 2017, 17, 8. [Google Scholar] [CrossRef]
- Gadelha, M.R.; Kasuki, L.; Lim, D.S.T.; Fleseriu, M. Systemic Complications of Acromegaly and the Impact of the Current Treatment Landscape: An Update. Endocr. Rev. 2019, 40, 268–332. [Google Scholar] [CrossRef] [Green Version]
- Battistone, M.F.; Miragaya, K.; Rogozinski, A.; Agüero, M.; Alfieri, A.; Ballarino, M.C.; Boero, L.; Danilowicz, K.; Diez, S.; Donoso, M.; et al. Increased risk of preneoplastic colonic lesions and colorectal carcinoma in acromegaly: Multicenter case-control study. Pituitary 2021, 24, 96–103. [Google Scholar] [CrossRef]
- Foltyn, W.; Kos-Kudla, B.; Strzelczyk, J.; Matyja, V.; Karpe, J.; Rudnik, A.; Marek, B.; Kajdaniuk, D.; Sieron, A.; Latos, W. Is there any relation between hyperinsulinemia, insulin resistance and colorectal lesions in patients with acromegaly? Neuro. Endocrinol. Lett. 2008, 29, 107–112. [Google Scholar]
- Jenkins, P.J.; Frajese, V.; Jones, A.M.; Camacho-Hubner, C.; Lowe, D.G.; Fairclough, P.D.; Chew, S.L.; Grossman, A.B.; Monson, J.P.; Besser, G.M. Insulin-like growth factor I and the development of colorectal neoplasia in acromegaly. J. Clin. Endocrinol. Metab. 2000, 85, 3218–3221. [Google Scholar] [CrossRef]
- Renehan, A.G.; O’Connell, J.; O’Halloran, D.; Shanahan, F.; Potten, C.S.; O’Dwyer, S.T.; Shalet, S.M. Acromegaly and colorectal cancer: A comprehensive review of epidemiology, biological mechanisms, and clinical implications. Horm. Metab. Res. 2003, 35, 712–725, Erratum in Horm. Metab. Res. 2004, 36, 70–71. [Google Scholar] [CrossRef]
- Dworakowska, D.; Grossman, A.B. Colonic Cancer and Acromegaly. Front. Endocrinol. 2019, 10, 390. [Google Scholar] [CrossRef]
- Parolin, M.; Dassie, F.; Russo, L.; Mazzocut, S.; Ferrata, M.; De Carlo, E.; Mioni, R.; Fallo, F.; Vettor, R.; Martini, C.; et al. Guidelines versus real life practice: The case of colonoscopy in acromegaly. Pituitary 2018, 21, 16–24. [Google Scholar] [CrossRef]
- Dworakowska, D.; Gueorguiev, M.; Kelly, P.; Monson, J.P.; Besser, G.M.; Chew, S.L.; Akker, S.A.; Drake, W.M.; Fairclough, P.D.; Grossman, A.B.; et al. Repeated colonoscopic screening of patients with acromegaly: 15-year experience identifies those at risk of new colonic neoplasia and allows for effective screening guidelines. Eur. J. Endocrinol. 2010, 163, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Fukuoka, H.; Iguchi, G.; Matsumoto, R.; Takahashi, M.; Nishizawa, H.; Suda, K.; Bando, H.; Takahashi, Y. The prevalence and associated factors of colorectal neoplasms in acromegaly: A single center based study. Pituitary 2015, 18, 343–351. [Google Scholar] [CrossRef]
- Rokkas, T.; Pistiolas, D.; Sechopoulos, P.; Margantinis, G.; Koukoulis, G. Risk of colorectal neoplasm in patients with acromegaly: A meta-analysis. World J. Gastroenterol. 2008, 14, 3484–3489. [Google Scholar] [CrossRef]
- Dutta, P.; Bhansali, A.; Vaiphei, K.; Dutta, U.; Ravi Kumar, P.; Masoodi, S.; Mukherjee, K.K.; Varma, A.; Kochhar, R. Colonic neoplasia in acromegaly: Increased proliferation or deceased apoptosis? Pituitary 2012, 15, 166–173. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wen, J.; Tian, T.; Lu, Z.; Wang, Y.; Wang, Z.; Wang, X.; Yang, Y. GLUT-1 overexpression as an unfavorable prognostic biomarker in patients with colorectal cancer. Oncotarget 2017, 8, 11788–11796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, M.; Samal, J.; Kandpal, M.; Singh, O.V.; Vivekanandan, P. The Warburg effect: Insights from the past decade. Pharmacol. Ther. 2013, 137, 318–330. [Google Scholar] [CrossRef]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, P.; Martel, F. Butyrate and colorectal cancer: The role of butyrate transport. Curr. Drug Metab. 2013, 14, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell. 2012, 48, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, P.; Martel, F. Regulation of colonic epithelial butyrate transport: Focus on colorectal cancer. Porto Biomed. J. 2016, 1, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, B.; Mirnezami, R.; Kinross, J.; Cloarec, O.; Keun, H.C.; Holmes, E.; Goldin, R.D.; Ziprin, P.; Darzi, A.; Nicholson, J.K. 1H HR-MAS NMR spectroscopy of tumor-induced local metabolic “field-effects” enables colorectal cancer staging and prognostication. J. Proteome Res. 2013, 12, 959–968. [Google Scholar] [CrossRef]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, H.; Liu, A.; Fang, C.; Hao, J.; Wang, Z. Lactate dehydrogenase A negatively regulated by miRNAs promotes aerobic glycolysis and is increased in colorectal cancer. Oncotarget 2015, 6, 19456–19468. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Simopoulos, C.; Polychronidis, A.; Sivridis, E. Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis 2005, 22, 25–30. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Trarbach, T.; Folprecht, G.; Shi, M.M.; Lebwohl, D.; Jalava, T.; Laurent, D.; et al. Prognostic and predictive role of lactate dehydrogenase 5 expression in colorectal cancer patients treated with PTK787/ZK 222584 (vatalanib) antiangiogenic therapy. Clin. Cancer Res. 2011, 17, 4892–4900. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, Y.; Hattori, K.; Taniguchi, K.; Tanaka, K.; Uchiyama, K.; Hirose, Y. Intratumoral heterogeneity of glutaminase and lactate dehydrogenase A protein expression in colorectal cancer. Oncol. Lett. 2020, 19, 2934–2942. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Franklin, C.C.; Chin, P.C.; Turner, J.T.; Kim, H.D. Insulin regulation of glucose metabolism in HT29 colonic adenocarcinoma cells: Activation of glycolysis without augmentation of glucose transport. Biochim. Biophys. Acta 1988, 972, 60–68. [Google Scholar] [CrossRef]
- MacDonald, R.S.; Thornton, W.H., Jr.; Bean, T.L. Insulin and IGE-1 receptors in a human intestinal adenocarcinoma cell line (CACO-2): Regulation of Na+ glucose transport across the brush border. J. Recept. Res. 1993, 13, 1093–1113. [Google Scholar] [CrossRef]
- Baghdiguian, S.; Verrier, B.; Gerard, C.; Fantini, J. Insulin like growth factor I is an autocrine regulator of human colon cancer cell differentiation and growth. Cancer Lett. 1992, 62, 23–33. [Google Scholar] [CrossRef]
- Nam, S.O.; Yotsumoto, F.; Miyata, K.; Fukagawa, S.; Yamada, H.; Kuroki, M.; Miyamoto, S. Warburg effect regulated by amphiregulin in the development of colorectal cancer. Cancer Med. 2015, 4, 575–587. [Google Scholar] [CrossRef]
- Wu, X.L.; Wang, L.K.; Yang, D.D.; Qu, M.; Yang, Y.J.; Guo, F.; Han, L.; Xue, J. Effects of Glut1 gene silencing on proliferation, differentiation, and apoptosis of colorectal cancer cells by targeting the TGF-β/PI3K-AKT-mTOR signaling pathway. J. Cell Biochem. 2018, 119, 2356–2367. [Google Scholar] [CrossRef]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wang, Y.; Ai, M.; Wang, H.; Duan, Z.; Wang, H.; Zhao, L.; Yu, J.; Ding, Y.; Wang, S. Long noncoding RNA CRNDE stabilized by hnRNPUL2 accelerates cell proliferation and migration in colorectal carcinoma via activating Ras/MAPK signaling pathways. Cell Death Dis. 2017, 8, e2862. [Google Scholar] [CrossRef]
- Wei, H.; Dong, C.; Shen, Z. Kallikrein-related peptidase (KLK10) cessation blunts colorectal cancer cell growth and glucose metabolism by regulating the PI3K/Akt/mTOR pathway. Neoplasma 2020, 67, 889–897. [Google Scholar] [CrossRef]
- Pechlivanis, S.; Wagner, K.; Chang-Claude, J.; Hoffmeister, M.; Brenner, H.; Försti, A. Polymorphisms in the insulin like growth factor 1 and IGF binding protein 3 genes and risk of colorectal cancer. Cancer Detect. Prev. 2007, 31, 408–416. [Google Scholar] [CrossRef]
- Karimi, K.; Mahmoudi, T.; Karimi, N.; Dolatmoradi, H.; Arkani, M.; Farahani, H.; Vahedi, M.; Parsimehr, E.; Dabiri, R.; Nobakht, H.; et al. Is there an association between variants in candidate insulin pathway genes IGF-I, IGFBP-3, INSR, and IRS2 and risk of colorectal cancer in the Iranian population? Asian Pac. J. Cancer Prev. 2013, 14, 5011–5016. [Google Scholar] [CrossRef] [Green Version]
- Stanilov, N.S.; Karakolev, I.A.; Deliysky, T.S.; Jovchev, J.P.; Stanilova, S.A. Association of insulin-like growth factor-I receptor polymorphism with colorectal cancer development. Mol. Biol. Rep. 2014, 41, 8099–8106. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Cui, G.; Tang, C.W.; Zhang, X.L.; Dai, C.; Xu, Y.Q.; Gong, H.; Xue, T.; Guo, H.H.; Bao, Y. Role of glucose metabolism related gene GLUT1 in the occurrence and prognosis of colorectal cancer. Oncotarget 2017, 8, 56850–56857. [Google Scholar] [CrossRef]
- de Kort, S.; Simons, C.C.J.M.; van den Brandt, P.A.; Janssen-Heijnen, M.L.G.; Sanduleanu, S.; Masclee, A.A.M.; Weijenberg, M.P. Diabetes mellitus, genetic variants in the insulin-like growth factor pathway and colorectal cancer risk. Int. J. Cancer 2019, 145, 1774–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.J.; Zheng, Z.J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adnan, Z. Sodium Glucose Co-transporter Inhibitors in Patients with Acromegaly and Diabetes. Trends Endocrinol. Metab. 2019, 30, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Soranna, D.; Scotti, L.; Zambon, A.; Bosetti, C.; Grassi, G.; Catapano, A.; La Vecchia, C.; Mancia, G.; Corrao, G. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: A meta-analysis. Oncologist 2012, 17, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thent, Z.C.; Zaidun, N.H.; Azmi, M.F.; Senin, M.I.; Haslan, H.; Salehuddin, R. Is Metformin a Therapeutic Paradigm for Colorectal Cancer: Insight into the Molecular Pathway? Curr. Drug Targets 2017, 18, 734–750. [Google Scholar] [CrossRef]
- Kheirandish, M.; Mahboobi, H.; Yazdanparast, M.; Kamal, W.; Kamal, M.A. Anti-cancer Effects of Metformin: Recent Evidences for its Role in Prevention and Treatment of Cancer. Curr. Drug Metab. 2018, 19, 793–797. [Google Scholar] [CrossRef]
- Kamarudin, M.N.A.; Sarker, M.M.R.; Zhou, J.R.; Parhar, I. Metformin in colorectal cancer: Molecular mechanism, preclinical and clinical aspects. J. Exp. Clin. Cancer Res. 2019, 38, 491. [Google Scholar] [CrossRef] [Green Version]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaafar, D.K.; Zaitone, S.A.; Moustafa, Y.M. Role of metformin in suppressing 1,2-dimethylhydrazine-induced colon cancer in diabetic and non-diabetic mice: Effect on tumor angiogenesis and cell proliferation. PLoS ONE 2014, 9, e100562. [Google Scholar] [CrossRef]
- Jain, D.; Chhoda, A.; Uribe, J. Effect of Insulin and Metformin Combination Treatment on Colon Adenoma and Advanced Adenoma Among DM II. J. Gastrointest. Cancer 2016, 47, 404–408. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Yu, M.; Xu, M.; Xiao, Y.; Ma, W.; Huang, L.; Li, X.; Ye, X. Berberine inhibits proliferation and induces G0/G1 phase arrest in colorectal cancer cells by downregulating IGF2BP3. Life Sci. 2020, 260, 118413. [Google Scholar] [CrossRef]
- Jia, Y.; Ma, Z.; Liu, X.; Zhou, W.; He, S.; Xu, X.; Ren, G.; Xu, G.; Tian, K. Metformin prevents DMH-induced colorectal cancer in diabetic rats by reversing the warburg effect. Cancer Med. 2015, 4, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Maximchik, P.; Abdrakhmanov, A.; Inozemtseva, E.; Tyurin-Kuzmin, P.A.; Zhivotovsky, B.; Gogvadze, V. 2-Deoxy-D-glucose has distinct and cell line-specific effects on the survival of different cancer cells upon antitumor drug treatment. FEBS J. 2018, 285, 4590–4601. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Chen, Q.; Gong, K.; Xu, X.; Xie, Y.; Zhang, W.; Cao, H.; Hu, T.; Hong, X.; Zhan, Y.Y. Berberine decelerates glucose metabolism via suppression of mTOR-dependent HIF-1α protein synthesis in colon cancer cells. Oncol. Rep. 2018, 39, 2436–2442. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Hu, X.; Xu, Y.; Yang, J.; Zong, L.; Wang, C.; Zhu, J.; Li, Z.; Lu, D. Berberine inhibits proliferation and migration of colorectal cancer cells by downregulation of GRP78. Anticancer Drugs 2020, 31, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Han, S.; Lei, K.; Chang, X.; Wang, K.; Li, Z.; Liu, J. Anti-Warburg effect of rosmarinic acid via miR-155 in colorectal carcinoma cells. Eur. J. Cancer Prev. 2016, 25, 481–489. [Google Scholar] [CrossRef]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprévote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc, L.; et al. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci. Rep. 2017, 7, 6945. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef]
- Hasanpourghadi, M.; Looi, C.Y.; Pandurangan, A.K.; Sethi, G.; Wong, W.F.; Mustafa, M.R. Phytometabolites Targeting the Warburg Effect in Cancer Cells: A Mechanistic Review. Curr. Drug Targets 2017, 18, 1086–1094. [Google Scholar] [CrossRef]
- Yao, Z.; Xie, F.; Li, M.; Liang, Z.; Xu, W.; Yang, J.; Liu, C.; Li, H.; Zhou, H.; Qu, L.H. Oridonin induces autophagy via inhibition of glucose metabolism in p53-mutated colorectal cancer cells. Cell Death Dis. 2017, 8, e2633. [Google Scholar] [CrossRef]
- Wu, M.; Li, H.; Liu, R.; Gao, X.; Zhang, M.; Liu, P.; Fu, Z.; Yang, J.; Zhang-Negrerie, D.; Gao, Q. Galactose conjugated platinum(II) complex targeting the Warburg effect for treatment of non-small cell lung cancer and colon cancer. Eur. J. Med. Chem. 2016, 110, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhao, Z.; Zhang, N.; Yang, Y.; Ma, L.; Feng, L.; Zhang, X.; Zuo, J.; Fan, Z.; Wang, Y.; et al. Transcriptional dysregulation of TRIM29 promotes colorectal cancer carcinogenesis via pyruvate kinase-mediated glucose metabolism. Aging (Albany N. Y.) 2021, 13, 5034–5054. [Google Scholar] [CrossRef]
- Wu, H.; Cui, M.; Li, C.; Li, H.; Dai, Y.; Cui, K.; Li, Z. Kaempferol Reverses Aerobic Glycolysis via miR-339-5p-Mediated PKM Alternative Splicing in Colon Cancer Cells. J. Agric. Food Chem. 2021, 69, 3060–3068. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A. Warburg effect, lactate dehydrogenase, and radio/chemo-therapy efficacy. Int. J. Radiat. Biol. 2019, 95, 408–426. [Google Scholar] [CrossRef] [PubMed]
- Passardi, A.; Scarpi, E.; Tamberi, S.; Cavanna, L.; Tassinari, D.; Fontana, A.; Pini, S.; Bernardini, I.; Accettura, C.; Ulivi, P.; et al. Impact of Pre-Treatment Lactate Dehydrogenase Levels on Prognosis and Bevacizumab Efficacy in Patients with Metastatic Colorectal Cancer. PLoS ONE 2015, 10, e0134732. [Google Scholar] [CrossRef] [PubMed]
- Mitov, M.I.; Harris, J.W.; Alstott, M.C.; Zaytseva, Y.Y.; Evers, B.M.; Butterfield, D.A. Temperature induces significant changes in both glycolytic reserve and mitochondrial spare respiratory capacity in colorectal cancer cell lines. Exp. Cell Res. 2017, 354, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.L.; Li, K.J.; Feng, J.X.; Liu, G.J.; Feng, Y.L. Blocking the IGF2BP1-promoted glucose metabolism of colon cancer cells via direct de-stabilizing mRNA of the LDHA enhances anticancer effects. Mol. Ther. Nucleic Acids. 2021, 23, 835–846. [Google Scholar] [CrossRef]
- Wang, K.; Huang, W.; Sang, X.; Wu, X.; Shan, Q.; Tang, D.; Xu, X.; Cao, G. Atractylenolide I inhibits colorectal cancer cell proliferation by affecting metabolism and stemness via AKT/mTOR signaling. Phytomedicine 2020, 68, 153191. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Liu, Z.; Guo, X.; Miao, Z.; Ma, S. Atractylenolide I Induces Apoptosis and Suppresses Glycolysis by Blocking the JAK2/STAT3 Signaling Pathway in Colorectal Cancer Cells. Front. Pharmacol. 2020, 11, 273. [Google Scholar] [CrossRef]
- Kitazawa, M.; Hatta, T.; Sasaki, Y.; Fukui, K.; Ogawa, K.; Fukuda, E.; Goshima, N.; Okita, N.; Yamada, Y.; Nakagama, H.; et al. Promotion of the Warburg effect is associated with poor benefit from adjuvant chemotherapy in colorectal cancer. Cancer Sci. 2020, 111, 658–666. [Google Scholar] [CrossRef] [Green Version]
- Woźniak, M.; Pastuch-Gawołek, G.; Makuch, S.; Wiśniewski, J.; Krenács, T.; Hamar, P.; Gamian, A.; Szeja, W.; Szkudlarek, D.; Krawczyk, M.; et al. In Vitro and In Vivo Efficacy of a Novel Glucose-Methotrexate Conjugate in Targeted Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 1748. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, X.; Zhou, Y.; Hu, Y. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol. Rep. 2012, 28, 1346–1352. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakagawa, Y.; Ito, Y.; Otsuki, Y.; Uno, B.; Uchiyama, K.; et al. MicroRNA-124 inhibits cancer cell growth through PTB1/PKM1/PKM2 feedback cascade in colorectal cancer. Cancer Lett. 2015, 363, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Sakai, M.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakayama, T.; Ueda, H.; Nakagawa, Y.; Ito, Y.; et al. PTBP1-associated microRNA-1 and -133b suppress the Warburg effect in colorectal tumors. Oncotarget 2016, 7, 18940–18952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Huang, Y.; Pan, Q.; Xiang, P.; Xie, N.; Yu, H. MicroRNA-98 Suppress Warburg Effect by Targeting HK2 in Colon Cancer Cells. Dig. Dis. Sci. 2017, 62, 660–668. [Google Scholar] [CrossRef]
- Pan, X.; Feng, J.; Zhu, Z.; Yao, L.; Ma, S.; Hao, B.; Zhang, G. A positive feedback loop between miR-181b and STAT3 that affects Warburg effect in colon cancer via regulating PIAS3 expression. J. Cell Mol. Med. 2018, 22, 5040–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Yang, P.; Amin, S.; Li, Z. A novel miR-206/hnRNPA1/PKM2 axis reshapes the Warburg effect to suppress colon cancer growth. Biochem. Biophys. Res. Commun. 2020, 531, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, Z.; Zou, K.; Cheng, Y.; Yang, M.; Chen, H.; Wang, H.; Zhao, J.; Chen, P.; He, L.; et al. MiR-1 suppresses tumor cell proliferation in colorectal cancer by inhibition of Smad3-mediated tumor glycolysis. Cell Death Dis. 2017, 8, e2761. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Cui, L.; Mei, Z.; Liu, M.; Zhang, D. miR-181a mediates metabolic shift in colon cancer cells via the PTEN/AKT pathway. FEBS Lett. 2014, 588, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Tan, F.; Yu, N.; Pei, H. STAT1 Inhibits MiR-181a Expression to Suppress Colorectal Cancer Cell Proliferation Through PTEN/Akt. J. Cell Biochem. 2017, 118, 3435–3443. [Google Scholar] [CrossRef]
- Li, A.; Qiu, M.; Zhou, H.; Wang, T.; Guo, W. PTEN, Insulin Resistance and Cancer. Curr. Pharm. Des. 2017, 23, 3667–3676. [Google Scholar] [CrossRef] [PubMed]
- Santasusagna, S.; Moreno, I.; Navarro, A.; Muñoz, C.; Martinez, F.; Hernández, R.; Castellano, J.J.; Monzo, M. miR-328 mediates a metabolic shift in colon cancer cells by targeting SLC2A1/GLUT1. Clin. Transl. Oncol. 2018, 20, 1161–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; Su, L.; Shan, J.; Zhu, C.; Liu, L.; Liu, C.; Xu, Y.; Yang, Z.; Bian, X.; Shao, J.; et al. IGF/STAT3/NANOG/Slug Signaling Axis Simultaneously Controls Epithelial-Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer. Stem Cells 2016, 34, 820–831. [Google Scholar] [CrossRef]
- Chen, Y.C.; Ou, M.C.; Fang, C.W.; Lee, T.H.; Tzeng, S.L. High Glucose Concentrations Negatively Regulate the IGF1R/Src/ERK Axis through the MicroRNA-9 in Colorectal Cancer. Cells 2019, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Zuo, S.; Wu, L.; Wang, Y.; Yuan, X. Long Non-coding RNA MEG3 Activated by Vitamin D Suppresses Glycolysis in Colorectal Cancer via Promoting c-Myc Degradation. Front. Oncol. 2020, 10, 274. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wei, M.; Wang, C.; Sun, D.; Liu, P.; Zhong, X.; Yu, W. Long noncoding RNA KCNQ1OT1 promotes colorectal carcinogenesis by enhancing aerobic glycolysis via hexokinase-2. Aging (Albany N. Y.) 2020, 12, 11685–11697. [Google Scholar] [CrossRef]
- Barrea, L.; Caprio, M.; Tuccinardi, D.; Moriconi, E.; Di Renzo, L.; Muscogiuri, G.; Colao, A.; Savastano, S. Obesity Programs of nutrition, Education, Research and Assessment (OPERA) group. Could ketogenic diet "starve" cancer? Emerging evidence. Crit. Rev. Food. Sci. Nutr. 2020, 4, 1–22. [Google Scholar] [CrossRef]
- Castejón, M.; Plaza, A.; Martinez-Romero, J.; Fernandez-Marcos, P.J.; Cabo, R.; Diaz-Ruiz, A. Energy Restriction and Colorectal Cancer: A Call for Additional Research. Nutrients 2020, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, R.L.; Kaiser, D.L. Effects of calorie restriction and weight loss on glucose and insulin levels in obese humans. J. Am. Coll. Nutr. 1985, 4, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Gabel, K.; Kroeger, C.M.; Trepanowski, J.F.; Hoddy, K.K.; Cienfuegos, S.; Kalam, F.; Varady, K.A. Differential Effects of Alternate-Day Fasting Versus Daily Calorie Restriction on Insulin Resistance. Obesity (Silver Spring) 2019, 27, 1443–1450. [Google Scholar] [CrossRef]
- Wong, M.H.; Holst, C.; Astrup, A.; Handjieva-Darlenska, T.; Jebb, S.A.; Kafatos, A.; Kunesova, M.; Larsen, T.M.; Martinez, J.A.; Pfeiffer, A.F.; et al. Caloric restriction induces changes in insulin and body weight measurements that are inversely associated with subsequent weight regain. PLoS ONE 2012, 7, e42858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmani, J.; Kord Varkaneh, H.; Clark, C.; Zand, H.; Bawadi, H.; Ryan, P.M.; Fatahi, S.; Zhang, Y. The influence of fasting and energy restricting diets on IGF-1 levels in humans: A systematic review and meta-analysis. Ageing Res. Rev. 2019, 53, 100910. [Google Scholar] [CrossRef] [PubMed]
- Vergati, M.; Krasniqi, E.; Monte, G.D.; Riondino, S.; Vallone, D.; Guadagni, F.; Ferroni, P.; Roselli, M. Ketogenic Diet and Other Dietary Intervention Strategies in the Treatment of Cancer. Curr. Med. Chem. 2017, 24, 1170–1185. [Google Scholar] [CrossRef]
- Pasanisi, P.; Gargano, G.; Gaetana Di Mauro, M.; Cortellini, M.; Casagrande, A.; Villarini, A.; Bruno, E.; Roveda, E.; Saibene, G.; Venturelli, E.; et al. A randomized controlled trial of Mediterranean diet and metformin to prevent age-related diseases in people with metabolic syndrome. Tumori 2018, 104, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Schmid, D.; Behrens, G.; Matthews, C.E.; Leitzmann, M.F. Physical Activity and Risk of Colon Cancer in Diabetic and Nondiabetic US Adults. Mayo Clin. Proc. 2016, 91, 1693–1705. [Google Scholar] [CrossRef]
- Sax, A.T.; Jenkins, D.G.; Devin, J.L.; Hughes, G.I.; Bolam, K.A.; Skinner, T.L. The insulin-like growth factor axis: A biological mechanism linking physical activity to colorectal cancer survival. Cancer Epidemiol. 2014, 38, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Oruç, Z.; Kaplan, M.A. Effect of exercise on colorectal cancer prevention and treatment. World J. Gastrointest. Oncol. 2019, 11, 348–366. [Google Scholar] [CrossRef]
- Berkovic, M.C.; Cigrovski, V.; Bilic-Curcic, I.; Mrzljak, A. What is the gut feeling telling us about physical activity in colorectal carcinogenesis? World J. Clin. Cases 2020, 8, 5844–5851. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslami, M.; Sadrifar, S.; Karbalaei, M.; Keikha, M.; Kobyliak, N.M.; Yousefi, B. Importance of the Microbiota Inhibitory Mechanism on the Warburg Effect in Colorectal Cancer Cells. J. Gastrointest. Cancer 2020, 51, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Curry, K.P.; Bultman, S.J. Microbial oncotarget: Bacterial-produced butyrate, chemoprevention and Warburg effect. Oncotarget 2013, 4, 182–183. [Google Scholar] [CrossRef] [Green Version]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| IGF Component in Serum (S) and Tissue (T) | Material and Methods | Risk/Incidence of Neoplastic Change/CRC | Serum/Local Tissue Level | Ref. | |
|---|---|---|---|---|---|
| IGF-1 peptide | S | 193 cases; 318 controls; ELISA | ↑CRC | ↑quintile vs. ↓quintile a (men) | [112] |
| 79 cases; 107 early-stage and 90 intermediate/late-stage adenomas; ELISA | ↑CRC and large or tubulovillous/villous adenoma; Δearly-stage adenoma | ↑tertile vs. ↓tertile (woman) | [115] | ||
| 75 cases; 146 controls; immunoradiometric assays | ΔCRC | ΔIGF-1 | [118] | ||
| cohort of 93,676 postmenopausal women; 438 incident cases; 816 random subcohort; ELISA | ↑CRC | free IGF-1 in multivariate models (woman) | [28] | ||
| 764 colon adenomas; 775 controls; ELISA | ↑colorectal adenoma | ↑IGF-1 | [113] | ||
| 73 colon and 410 rectal cancers; 650 controls; 120 post-operation colon and 211 rectal cancers; ELISA | CRC vs. control and post-operation cancers | ↑insulin, ↑IGF-1, ↑IGF-1/IGFBP-3 ratio | [114] | ||
| 95 cases; 48 controls; ELISA | CRC vs. control | ↑IGF-1, ↑IGFBP-2, ↑VEGF a | [116] | ||
| T | 10 cases; 10 controls; specific immunoassays | CRC vs. control | (+) in small but equal amounts in normal and malignant tissue | [125] | |
| 713 cases; IHC | CRC | (+) in 7.5%;  tumor stage (pT1/pT2) and proliferation activity a tumor stage (pT1/pT2) and proliferation activity a | [120] | ||
| 210 cases; IHC | CRC | (+) in 80% CRC; tumor size a; depth of invasion a | [122] | ||
| 10 cases; 10 controls; IHC | CRC and control pairs | (−) in all CRCs and controls | [124] | ||
| 28 cases; 28 controls; IHC | CRC and control pairs | (+) in 50% CRC; (+) in 39% controls | [121] | ||
| IGF-1 mRNA | T | 10 cases; 10 controls; Northern blot | CRC vs. control pairs | (+) IGF-1 in CRC and controls; ΔCRC vs. controls | [124] |
| 10 cases; 10 controls; hybridisation RNase protection assay | CRC and control | (−) IGF-1 mRNA | [125] | ||
| 90 cases (63 adenomas and 27 submucosal pT1 cancers); 90 controls; semiquantitative RT-PCR | CRC and control pairs | (+) in 54.4% cases; histopathology; (−) in controls or only faintly detected | [123] | ||
| 28 cases; 28 controls; real-time PCR | CRC vs. control pairs | ↓total IGF-1 mRNA and all mRNA isoforms a | [121] | ||
| 202 cases; 202 controls; RT-PCR | CRC vs. control | ↓IGF-1 mRNA a | [126] | ||
| IGF-1R peptide/mRNA | T | 713 cases; IHC | CRC | (+) in 99.6% cases (peptide) | [120] |
| 90 cases (63 adenomas and 27 submucosal pT1 cancers); 90 controls; RT-PCR | CRC and control pairs | (+) in 37.8% cases; (−) in controls or only faintly detected (mRNA) | [123] | ||
| 210 cases; IHC | CRC | (+) in 66% cases (peptide); tumor size a; depth of invasion | [122] | ||
| 202 cases; 202 controls; RT-PCR | CRC vs. control | ↑IGF-1R mRNA a | [126] | ||
| IGFBP-3 | S | 193 cases; 318 controls; ELISA | ↑CRC | ↓IGFBP-3 a (men) | [112] |
| 79 cancers, 107 early-stage and 90 intermediate/late-stage adenomas; ELISA | ↑CRC and large or tubulovillous/villous colorectal adenoma; Δearly-stage adenoma | ↓IGFBP-3 a (woman) | [115] | ||
| 75 cases; 146 controls; immunoradiometric assays | ↑CRC | ↑quintile vs. ↓quantile a | [118] | ||
| cohort of 93,676 postmenopausal women; 438 incident cases; 816 random subcohort; ELISA | ΔCRC | ΔIGFBP-3 | [28] | ||
| 764 colon adenomas; 775 controls; ELISA | Δadenoma | ΔIGFBP-3 | [113] | ||
| 73 colon and 410 rectal cancers, 650 controls, 120 post-operation colon and 211 rectal cancers; ELISA | CRC vs. control and post-operation cancers | ↓IGFBP-3 a | [114] | ||
| 95 cases; 48 controls; ELISA | CRC vs. control | ↓IGFBP-3 a | [116] | ||
| T | 10 cases; 10 controls; IHC | CRC and control pairs | (+) in 7/10 CRC and controls | [124] | |
| 202 cases; 202 controls; RT-PCR | CRC vs. control | ΔmRNA; lymph node metastasis; ♦ ↑poor 5-year overall survival | [126] | ||
—significant correlation between IGF component and clinical data; ♦—association between IGF component and CRC prognosis; CRC—colorectal cancer; ELISA-enzyme-linked immunosorbent assay; IGF-1-insulin-like growth factor 1; IGFBP-3-IGF binding protein 3; IGF-1R-IGF receptor type I; IHC-immunohistochemistry; RT-PCR-reverse transcription polymerase chain reaction.| Research Model | IGF-1 | Insulin | Glucose Metabolic Effects | Ref. |
|---|---|---|---|---|
| LID mice | a complete abrogation of liver IGF-1 mRNA; ↓↓(~75%) in cIGF-1 | 4-fold↑levels; muscle-specific insulin insensitivity | (i) glucose levels were normal vs. control; (ii) abnormal glucose clearance after insulin injection | [8] |
| LID mice; rhIGF-1 (1 mg/kg); ip for 20 days | ↓levels and ↑insulin sensitivity | ↓glucose levels to 40% of basal levels | ||
| MID mice | lower mIGF-1; ∼40% ↓of cIGF-1 in bMID mice at 4-wk-old mice | ↑levels in 8-wk-old male bMID, did not change in 12-wk-old bMID, and ↓in 16-wk-old bMID; ↓in fed aMID mice and no response upon food retention; HOMA-IR > 4-fold ↑in male aMID mice | (i) mIgf-1 deletion in male aMID mice alters glucose handling and ↑GLUT4 levels; (iii) mIGF-1 modulates anabolism and metabolism in an age-dependent manner; (ii) ↓mIGF-1 progressively disrupts glucose homeostasis in male mice | [80] |
| WT mice; IGF-1 (1 µg/mL); icv | ↑↑levels; ↑insulin sensitivity | (i) ↑food intake; (ii) ↓blood glucose levels; (iii) improves glucose tolerance | [146] | |
| WT mice; anti-IGF-1 Ab; icv | ↓levels; normal insulin sensitivity | (i) ↓appetite/food intake; (ii) ↑glucose levels; (iii) normal glucose tolerance | ||
| WT mice; 1011 GC/mouse of the AAV-Igf1 into ARC | ↑↑expression in ARC of the hypothalamus | ↑↑levels; ↑insulin sensitivity | (i) ↑appetite but unchanged body weight; (ii) ↓blood glucose levels; (iii) improves glucose tolerance | |
| WT mice (igf-1(+/+); untreated Hz mice (Hz, igf-1(+/−) and Hz, igf-1(+/−) mice treated with IGF-1 (Hz + IGF-1); C | ↓cIGF-1 vs. C; ↓igf-1 liver expression in untreated Hz groups vs. WT | ↑levels of glucose, triglycerides and cholesterol in the untreated Hz group as compared to both C and Hz + IGF-1 groups | [13] | |
| healthy adults; rhIGF-1 (100 µg/kg); iv | ↑level 15 min after injection, of which 80% was free IGF-1 (the highest level of free IGF-1 was 350 ng/mL) | (i) the acute hypoglycemia; the lowest blood glucose levels were reached after 30 min: 1.98 ± 0.44 mmol/L; (ii) on a molar basis, was only 6% as potent as insulin in the production of hypoglycemia | [100] | |
| healthy adults; insulin (0.15 IU/kg); iv | (i) the lowest blood glucose levels were reached after 30 min: 1.78 ± 0.29; (ii) inhibits lipolysis more effectively than IGF-1 | |||
| healthy adults; rhIGF-1 (20 µg/kg per h); sc; 6 days | ↑levels within 2–4 h after starting the infusion, and reached levels of 700 ng/mL after 13–14 h | (i) all fasting values before, during, and after the infusion remained within normal limits; (ii) ↓insulin secretion | Blood glucose remained within normal limits (between 3.7 and 4.4 mmol/L) throughout the study | [169] |
| healthy adults; high (30 µg/kg) and low (5 µg/kg) doses of rhIGF-1 iv per h; high (23 nmol/kg), and low (0.04 nmol/kg) doses of insulin iv per h | ↑total IGF-1 during infusion to 360% of baseline level at the end of high doses and to 150% after low doses | ↑levels during high and low insulin doses 5.6- and 1.6-fold above baseline values, whereas they ↓by 25 ± 5 and 22 ± 4% during high and low IGF-1 doses, respectively | (i) glucose rate of disappearance ↑from baseline by 239 ± 16% with high IGF-1 dose vs. 197 ± 18% with insulin iv; (ii) hepatic glucose ↓production by 37 ± 6% during high dose IGF-1 vs. 89 ± 13% during insulin iv | [103] |
| healthy adults; rhIGF-1 (7 and 14 µg/kg); iv per h during standard OGTT and MTT, respectively | ↑total and free cIGF-1 within 10 h after I infusion; on day 2, cIGF-1 were 3.9 and 4.4 times (total), and 1.8 and 4.1 times (free), respectively, above starting levels | (i) ↓insulin by direct suppression of its secretion; (ii) ↓insulin/glucose-ratio; (iii) ↑insulin sensitivity | (i) glucose tolerance remained unchanged in the face of ↓insulin | [104] |
| nondiabetic subjects with a wide range of BMI | (i) cIGF-1 negatively correlates with IVGTT-derived and OGTT-derived indexes I- and II phase insulin secretion; (ii) ↓cIGF-1 is associated with ↓insulin sensitivity | (i) cIGF-1 positively correlates with glucose disposal; (ii)low cIGF-1 is associated with obesity-related changes, MetS, glucose intolerance, and the development of DM II | [15] | |
| rats; STZ-induced DM | ↓cIGF-1; ↓↓IGF-1a/b mRNAs in liver, kidney, and lung tissues; treatment with insulin for 1 wk restored both IGF-1 mRNAs content toward that present in tissues of nondiabetic rats | [167] | ||
| DM I subjects (age 13–24 yrs) rhIGF-1/IGFBP-3 complex; 2 days; sc; two groups and placebo | cIGF-1 levels were in the physiological range | ↑insulin sensitivity following the two highest doses of rhIGF-1/IGFBP-3, whereas the lower doses had little effect on insulin sensitivity | Enhances glucose metabolism by controlling both endogenous glucose output and peripheral glucose uptake | [105] |
| (i) nondiabetic subjects; (ii) subjects with impaired glucose tolerance; (iii) DM II subjects | ↓cIGF-1 in subjects with MetS vs. subjects without MetS | (i) cIGF-1 positively correlates with HOMA-S; (ii) cIGF-1 independently correlates with insulin sensitivity | cIGF-1 levels are independently related with other components of MetS (impaired glucose regulation) | [12] |
| DM I subjects; rhIGF-1 (40 µg/kg); sc; basal insulin infusion iv and a hyperinsulinemic clamp | ↑cIGF-1 with max 4 h after the injection (398.2 ± 34.9 ng/L) | ↑level during the hyperinsulinemic euglycemic clamp | (i) ↓hepatic glucose production rate; (ii) ↑peripheral glucose uptake; (iii) direct effect on glucose and protein metabolism and acts together with insulin | [64] |
| obese subjects with DM II and with IR; rhIGF-1 (100 µg/kg); sc for 6 wks | ↑cIGF-1 was accompanied by a ↑IGFBP-2, slight ↓IGFBP-3, and ↑IGFBP-1 | ↓mean levels from 108.0 to 57.0 pmol/L during the modal day measurements and from 97.2 to 72.0 pmol/L during the MMT | (i) ↓blood glucose; (ii) ↑insulin sensitivity; (iii) improves glycemic control in DM II were associated with ↓insulin levels | [106] |
| Non-Coding RNA | Research Model | Mechanism of Change in Function | Ref. | |
|---|---|---|---|---|
| miRNAs | miR-124, miR-137, miR-340 | HCT116, DLD1, SW480 and HT29 cells; CRC tissues | (i) switch PKM gene expression from PKM2 to PKM1; (ii) ↓glycolysis rate, but ↑the glucose flux into oxidative phosphorylation | [246] |
| miR-124 | CRC cells; xenografted mice; CRC tissues | (i) ↓in CRC and adenoma tissues vs. adjacent tissue (ii) acts as a tumor-suppressor; (iii) ↑apoptosis and/or autophagic survival; (iv) targets PTB1 through the switching of PKM isoform expression from PKM2 to PKM1 | [247] | |
| miR-181a | HCT15 and HCT116 cells; CRC tissues | (i) ↑in CRC tissue; (ii) ↑cell proliferation through ↑glycolysis; (iii) suppressed PTEN expression by targeting its 3′-UTR, thus resulting in ↑Akt phosphorylation; (iv) causes an ↑lactate production and ↑cell proliferation through the PTEN/Akt pathway | [253] | |
| LoVo and SW480 cells | (i) ↑cell proliferation through PTEN; (ii) ↑PTEN in response to STAT1 overexpression or miR-181a inhibition; (iii) ↓PTEN in response to STAT1 knockdown or miR-181a overexpression | [254] | ||
| miR-1, miR-133b | DLD-1 cells and WiDr cells; xenografted mice; CRC tissues | (i) ↓in CRC and adenomas vs. C tissue; ↑in C tissue except muscle; (ii) induces growth suppression and autophagic cell death through the switching from PKM2 to PKM1 by silencing PTBP1 expression; (iii) ↑↑PTBP1 expression in CRC and adenomas | [249] | |
| miR-1 | HCT116, SW480, SW620, HT-29 cells; mice | (i) suppresses aerobic glycolysis and tumor cell proliferation via inactivation of Smad3 and targeting HIF-1α, leading to ↓HK II and ↓MCT4 expression (ii) Smad3 was central to the effects of miR-1 in CRC | [252] | |
| miR-98 | SW480 and HCT116 cells; CRC tissues | (i) ↓in CRC vs. C tissue; (ii) inhibits glycolysis by targeting HK II; (iii) negatively correlates with HK II expression in CRC tissues | [249] | |
| miR-181b (miR-181b-5p) | HCT116, HT-29, HEK-293T cells; xenografted mice | (i) is a direct regulator of PIAS3; (ii) promotes the Warburg effect and the growth of colon cancer xenografts; (iii) interacts with STAT3 phosphorylation in a positive feedback loop in CRC cells via regulating PIAS3 expression | [250] | |
| miR-206 | CRC cells | (i) ↓in CRC vs. C tissue; (ii) negative correlation with S, and inverse correlation with OS; (iii) causes ↓the cell proliferation, glucose consumption and lactate production; (iii) overexpression induces switching from PKM2 to PKM1; (iv) hnRNPA1 is a direct target of this miR to suppress PKM2 expression | [251] | |
| miR-34a, miR-34c, miR-369-3p, miR-374a, miR-4524a/b | HCT116, HCT15, HT29, Panc-1, Bxpc-3, CFPAC-1 cells; TMA with CRC; tumor bearing mice | (i) target LDHA and regulate glycolysis (ii) negatively correlates with LDHA expression in CRC tissues; (iii) a genetic loci newly associated with ↑CRC progression, rs18407893 at 11p15.4, which maps to the seed sequence recognized by miR-374a | [196] | |
| miR-328 | LOVO and SW480 cells; CRC tissues | (i) ↓in CRC vs. C tisuse; (ii) directly targets SLC2A1 3′-UTR; (iii) inhibits SLC2A1 and regulates GLUT1-mediated glycolytic activity in cancer cells | [256] | |
| miR-9 | SW480 and SW620 cells; CRC tissues | (i) ↓in CRC with hyperglycemia and with high levels of CEA; (ii) causes ↓IGF-1R/Src signaling and downstream cyclin B1 and N-cadherin, but ↑E-cadherin; (iii) high glucose level promoted cell proliferation, migration, and invasion ability of the cells, ↑G1 population, and EMT protein expression | [258] | |
| lncRNAs | CRNDE transcripts | HCT116, HT29, LS174T cells | (i) regulate genes involved in glucose and lipid metabolism; (ii) promote the metabolic changes by which cancer cells switch to aerobic glycolysis; (iii) are regulated by insulin/IGFs; (iv) downstream PI3K/Akt/mTOR and Raf/MAPK pathways repress CRNDE nuclear transcripts | [33] |
| MEG3 | DLD-1 and RKO cells; CRC tissues | (i) overexpression causes ↓glycolysis, and ↓lactate production in CRC cells; (ii) ↑degradates of c-Myc and ↓c-Myc target genes such as LDHA, PKM2 and HK II; (iii) can be activated by vit. D and VDR; (iv) vit. D-activated MEG3 causes ↓aerobic glycolysis in CRC cells via degradation of c-Myc | [259] | |
| KCNQ1OT1 | SW48, LoVo, HCT116, SW620, HT-29, RKO cells; CRC tissues | (i) ↑in CRC vs. C tissues; (ii) ↑expression correlates with poorer prognosis in patients; (iii) ↑CRC cell proliferation by ↑aerobic glycolysis; (iv) directly binds to HK II; (v)correlates positively with HK II expression and prognosis in CRC | [260] | |
| Targeted Agent | Model of the Study | Results | Role in CRC | Ref. |
|---|---|---|---|---|
| PTK787/ZK 222,584 (vatalanib) | CRC tissues; LDH serum; IHC method | (i) ↑LDH5 related to poor PFS only in the placebo group; (ii) vatalanib improved response and PFS in this group | predicting the response to CTX | [198] |
| CTX + bevacizumab and CTX only; NCT01878422 | mCRC patients | in patients with ↑LDH, the addition of bevacizumab to CTX led to a significant ↓in the rate of progressive disease and to a prolonged PFS | phase III prospective multicentre randomized ITACa | [239] |
| GLU-MTX | DLD-1 and SW-480 cells | (i) ↓cell viability (DLD-1); (ii) 17-fold ↑uptake of GLU-MTX in tumor cells vs. MTX (SW-480); (iii) cleavable linkage allows intracellular MTX release after selective uptake through GLUT1 | may offer a better tumor selectivity, growth inhibition at reduced toxicity | [245] |
| 2-DG | HTC116 and RKO cells | (i) triggers ER stress; (ii) in HCT116 cells ER stress stimulates autophagy | preventive/prospective | [227] |
| Gal-Pt; oxaliplatin | xenograft tumor model; HT-29 cells | (i) ↑therapeutic index by over 30-fold compared to cisplatin and oxaliplatin; (ii) the uptake of Gal-Pt was regulated by glucose transporters | preventive/prospective | [237] |
| TRIM29 | SW480 and HT29 cells; CRC tissues | (i) ↑in CRC vs. control; (ii) associated with poor clinical outcome; (iii) promotes the malignant phenotype in vitro and in vivo; (iv) promotes mainly PKM1 degradation; (v)directly targets PKM1 to ↓PKM1/PKM2 ratio | preventive/prospective | [236] |
| Resweratrol | Caco2 and HTC116 cells | (i) ↑PDH activity; (ii) ↑oxidative capacities and ↓glycolysis, in association with a ↓pentose phosphate activity and an ↑ATP production | preventive/prospective | [231] |
| Berberine | SW480 and HT-29 cells | (i) ↓cell proliferation and migration; (ii) ↑cell apoptosis, in a dose-dependent manner; (iii) ↓expression of GRP78 | preventive/prospective | [229] |
| HCT116 and KM12C cells | ↓glucose uptake and the transcription of GLUT1, LDHA and HK II genes | preventive/prospective | [228] | |
| bioinformatical analysis | (i) ↓cell proliferation and ↑G0/G1 phase arrest in CRC cells by ↓IGF2BP-3; (ii) knockdown of IGF2BP-3 could suppress the PI3K/Akt pathway to ↓cell proliferation | preventive/prospective | [88] | |
| Rosmarinic acid | CRC cells | (i) ↓glucose consumption and lactate generation; (ii) inhibits expression of HIF-1α; (iii) ↓the cytokines and miRNAs related to inflammation | preventive/prospective | [230] |
| Kaempferol | HCT116 and DLD1 cells | (i) ↓proliferation of cells, delayed G1 phase progression and ↑apoptosis; (ii) impaires glucose consumption, which causes ↓lactic acid accumulation and ATP production; (iii) promotes the expression of miR-339-5p with hnRNPA1 and PTBP1 as two direct targets | preventive/prospective | [237] |
| Atractylenolide I | COLO205 and HCT116 cells; mouse xenograft model | (i) inhibits invasion of cells by ↑apoptosis; (ii) alters glucose metabolism; (iii) suppresses stem-like traits; (iv) ↓Akt/mTOR; (v)↓tumor weight and volume | preventive/prospective | [242] |
| HCT116 and SW480 cells; male BALB/c nude mice injected with HCT116 cells | (i) ↓cell viability and colony formation; (ii) ↑cell apoptosis (iii) ↓cell glycolysis; (iv) inhibits STAT3/JAK2 activation | preventive/prospective | [243] | |
| Vitamin C | KRAS Mut CRC tissues; SW480, LoVo (KRAS Mut, G12V and G13D cells) and HCEC (KRAS WT) cells | (i) inhibits ERK 1/2 and PKM2 phosphorylation; (ii) ↓GLUT-1 and PKM2-PTBP dependent protein expression | preventive/prospective | [232] |
| Metformin (MET) | DMH-induced CRC in diabetic SD rats; LoVo and HT-29 cells | (i) inhibits the formation of ACF/tumors; (ii) inhibits cell growth and ↓the imbalance in the expression of the enzymes involved in glycolysis and the TCA cycle | preventive/prospective | [224] |
| HCT116 p53+/+ and p53−/− cells; both HCT116 cells inoculated of nude mice | (i) ↓the tumor growth of xenografts; (ii) ↓mitochondrial electron transport; (iii) ↑p53-dependent autophagy; (iv) ↑a metabolic conversion that p53−/− cells are unable to do | preventive/prospective | [220] | |
| MET; oxaliplatin; MET + oxaplatin | DMH-induced CRC in diabetic and non-diabetic mice | (i) ↑in angiogenic and cell proliferation markers; (ii) greater immunostaining for IGF-1R and CD34 in the colon of diabetic vs. non-diabetic mice | preventive/prospective | [221] |
| NCT02960711 (MET + moderate physical activity + Mediterranean-macrobiotic diet) | both sex with MetS; MET (1700 mg/day) + moderate PA, placebo + moderate PA, MET alone, and placebo | The Me.Me.Me. trial is ongoing. No patient has completed the 5 years of follow-up | preventive/prospective; phase III randomized controlled trial | [268] |
| Low- or high-fiber diets | BALB/c inbred mice associated with 4 commensal +/− the butyrate-producing B. fibrisolvens | A high-fiber diet protects against CRC tumors in a microbiota- and butyrate-dependent manner | preventive/prospective | [276] |
| Hyperthermia (HT) | SW480, HCT116, and Pt. 93 cells at 32 °C, 37 °C and 42 °C | Provides valuable insights for the metabolic and bioenergetic changes of CRC cells under hypothermia and HT conditions | preventive/prospective | [240] |
| HT resistant (HTR) LoVo cells | (i) ↑IGF2BP-1 in HTR cells vs. parental cells; (ii) LDHA mRNA was identified as an IGF2BP-1 direct target; (iii) inhibiting the IGF2BP-1-promoted glycolysis causes sensitisation of CRC cells to HT treatment | preventive/prospective | [241] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasprzak, A. Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6434. https://doi.org/10.3390/ijms22126434
Kasprzak A. Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. International Journal of Molecular Sciences. 2021; 22(12):6434. https://doi.org/10.3390/ijms22126434
Chicago/Turabian StyleKasprzak, Aldona. 2021. "Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer" International Journal of Molecular Sciences 22, no. 12: 6434. https://doi.org/10.3390/ijms22126434
APA StyleKasprzak, A. (2021). Insulin-Like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. International Journal of Molecular Sciences, 22(12), 6434. https://doi.org/10.3390/ijms22126434