
The Role of Hypoxic Bone Marrow Microenvironment in Acute Myeloid Leukemia and Future Therapeutic Opportunities

 ,
, 
Abstract
:1. Introduction
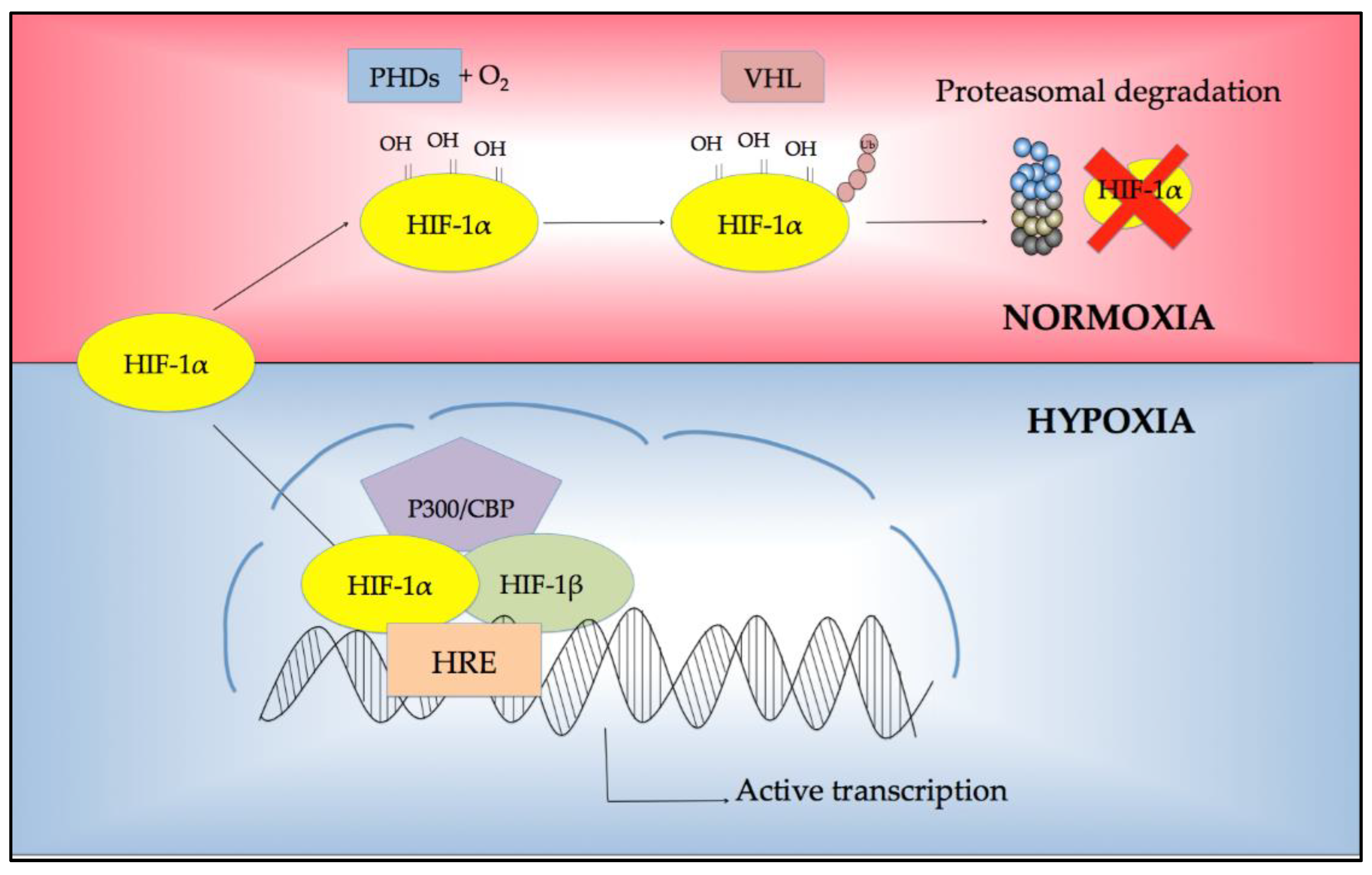
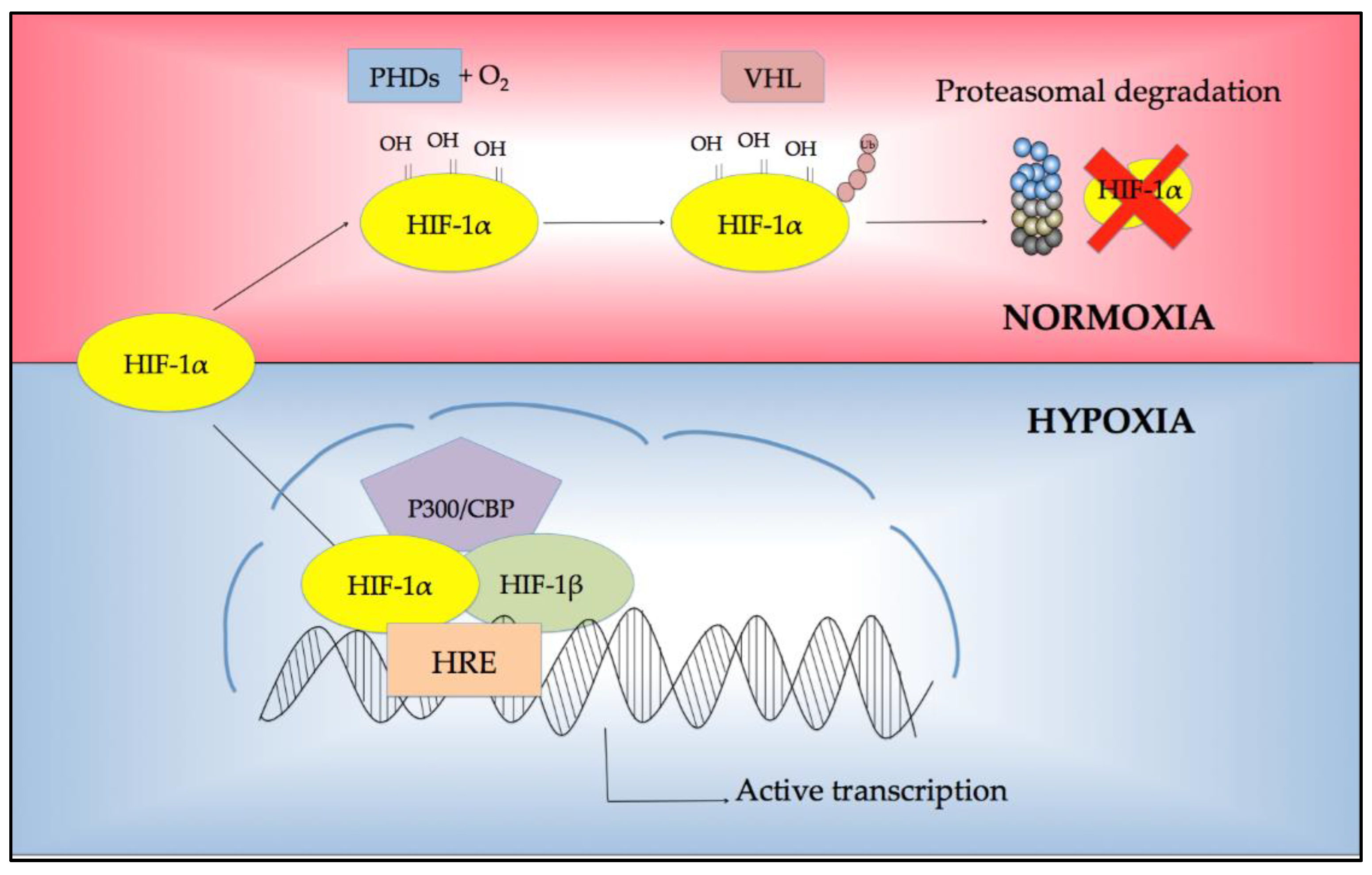
2. Regulation of Response to Hypoxia
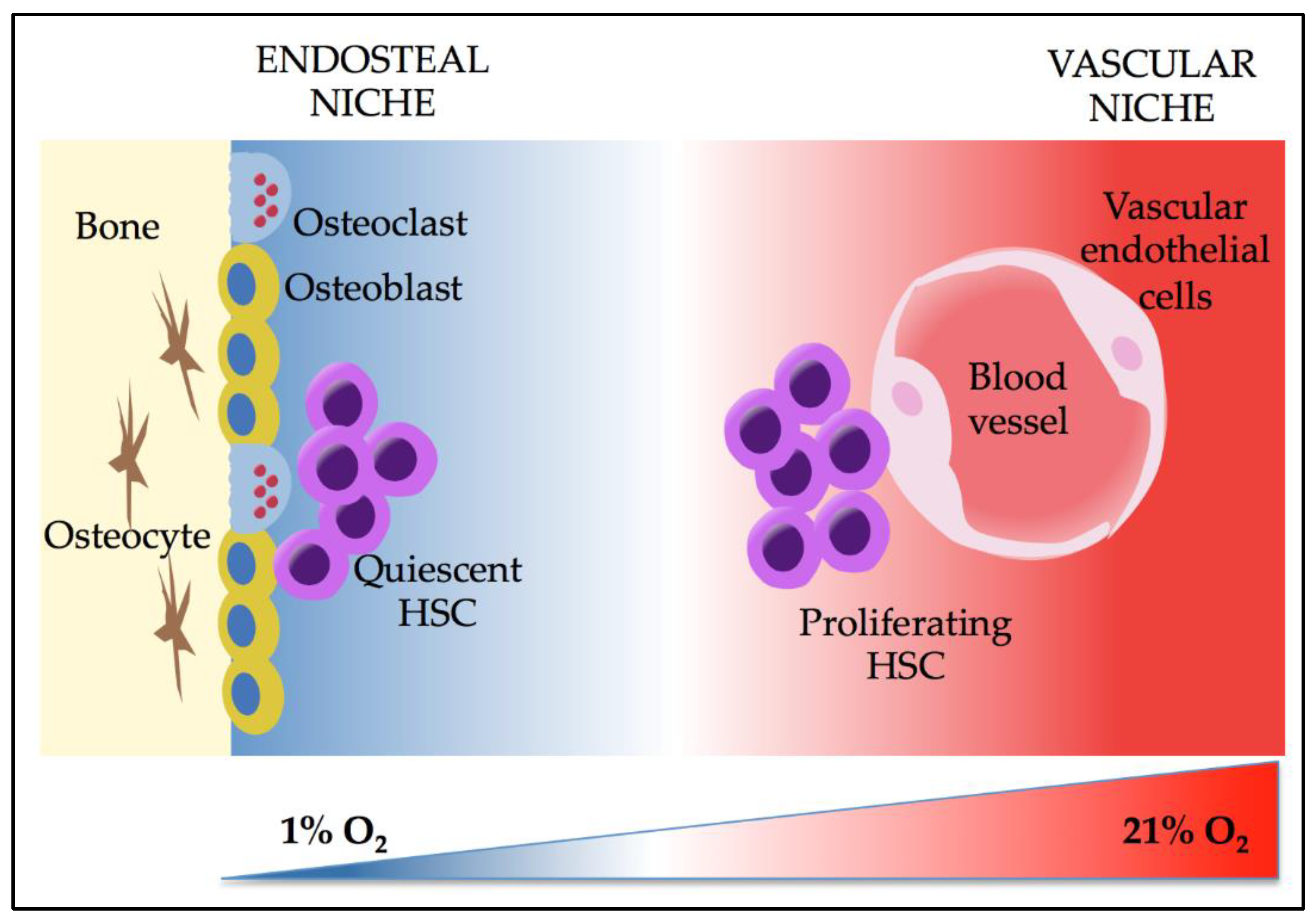
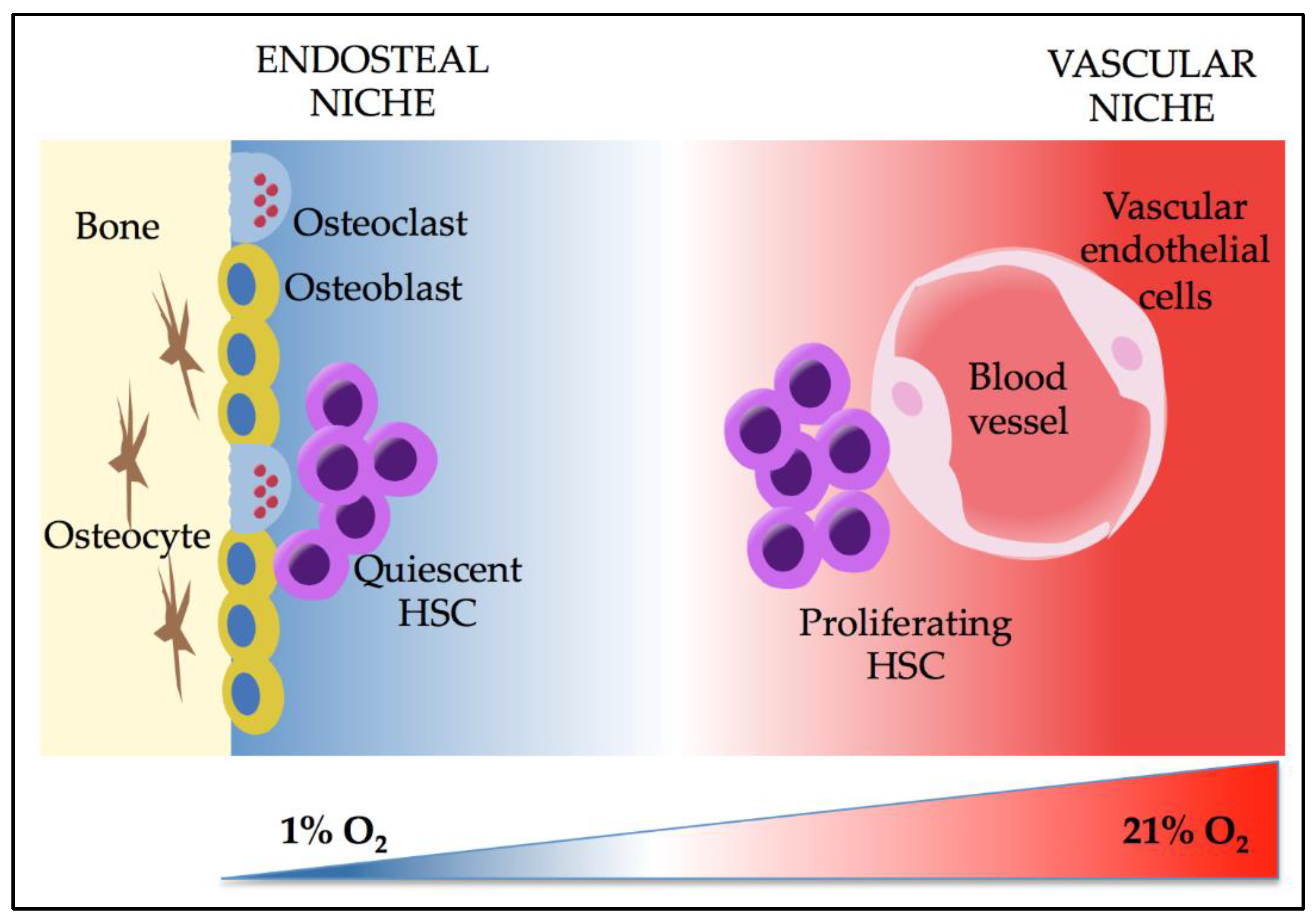
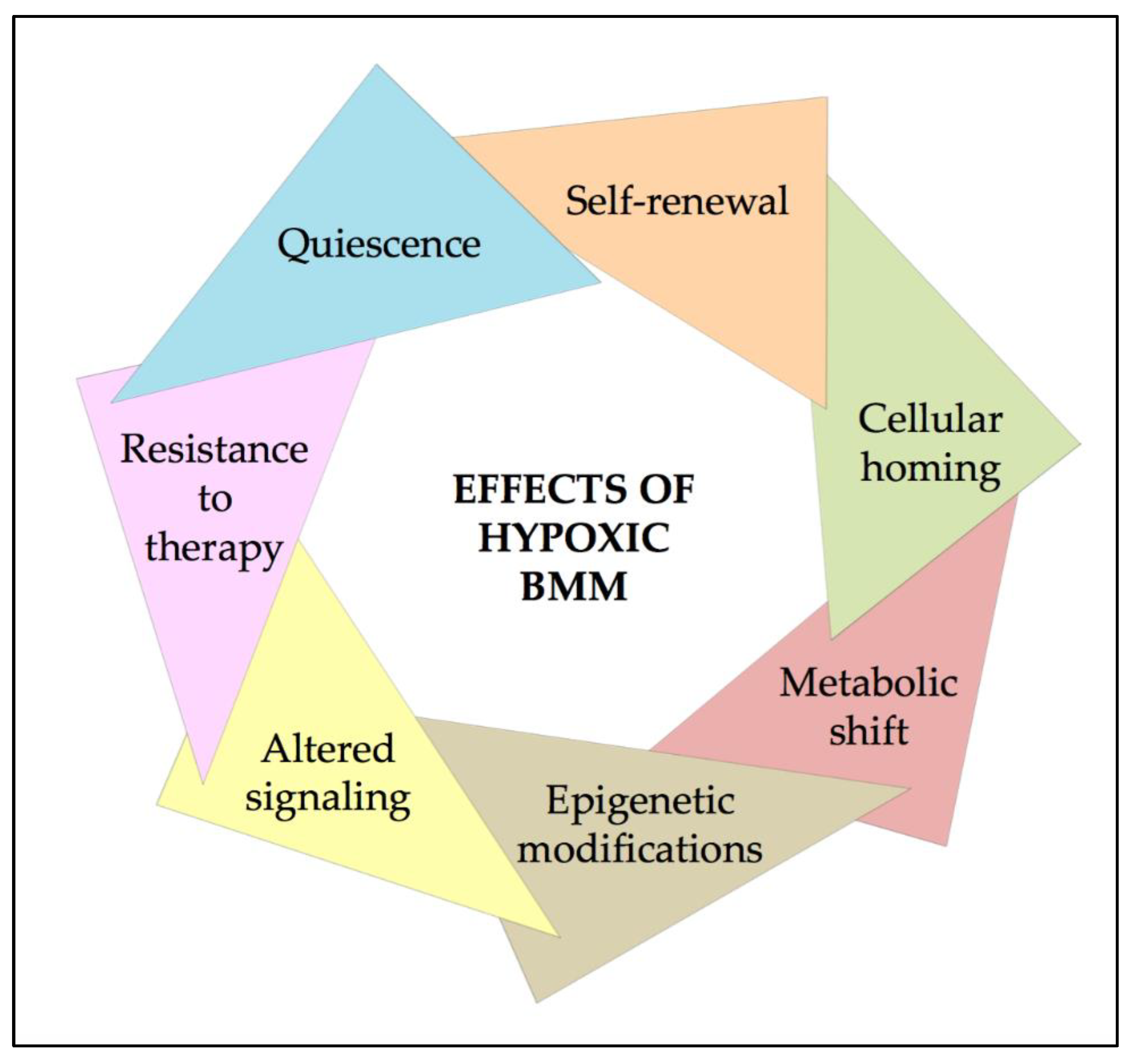
3. Effects of Hypoxia and HIFs on HSCs
4. HIFs in AML
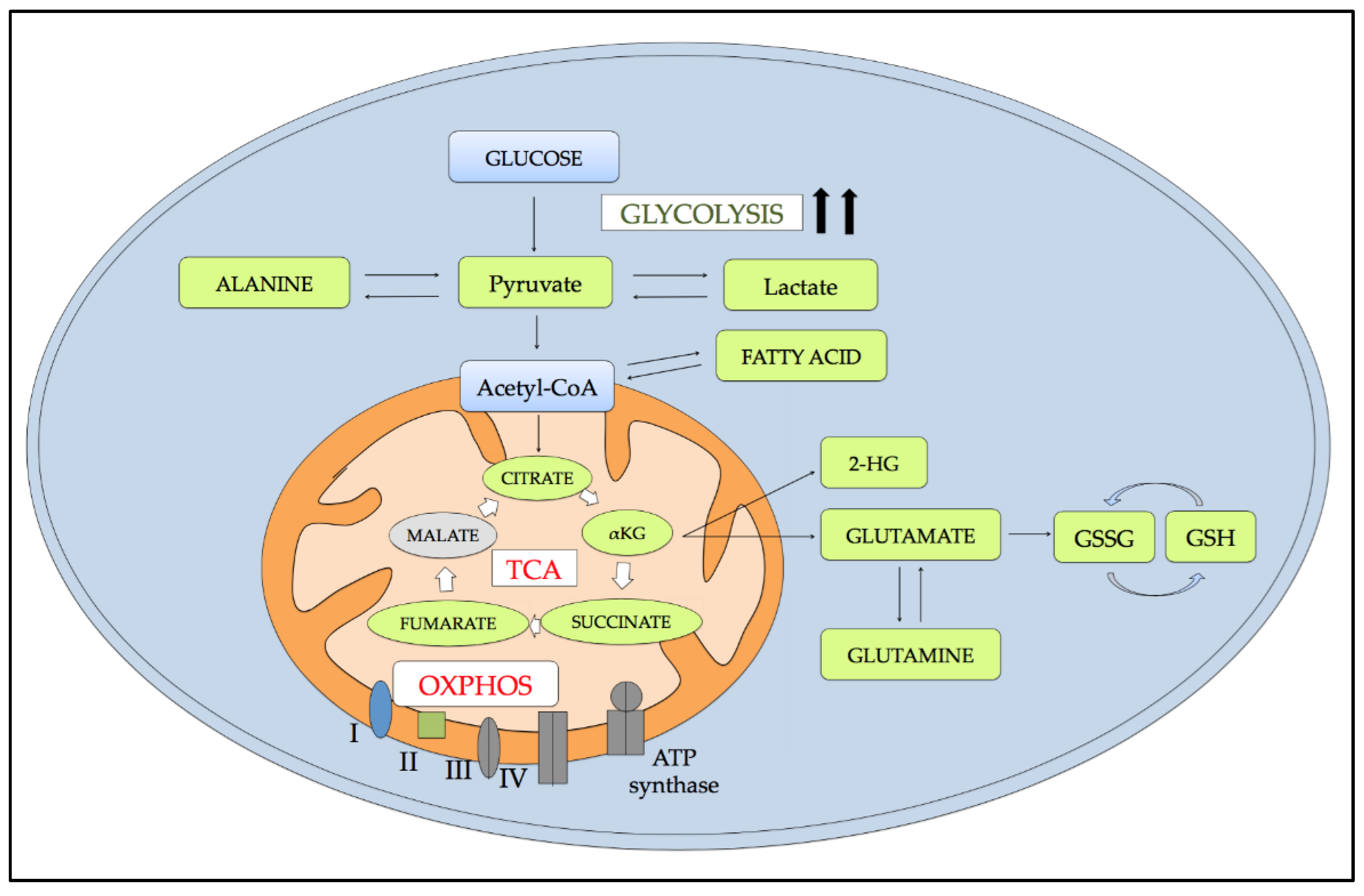
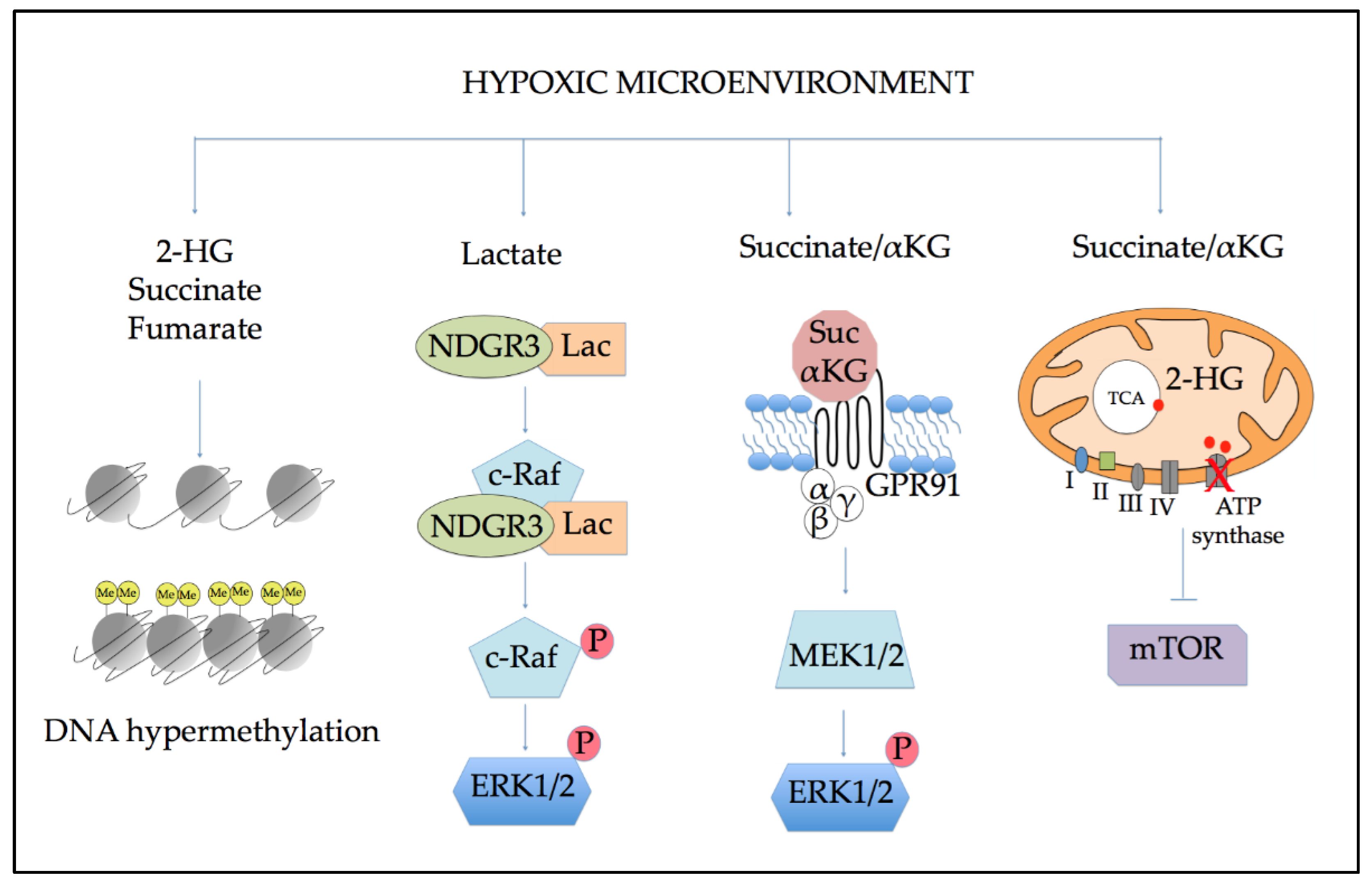
5. Metabolic Alterations Induced by Hypoxia
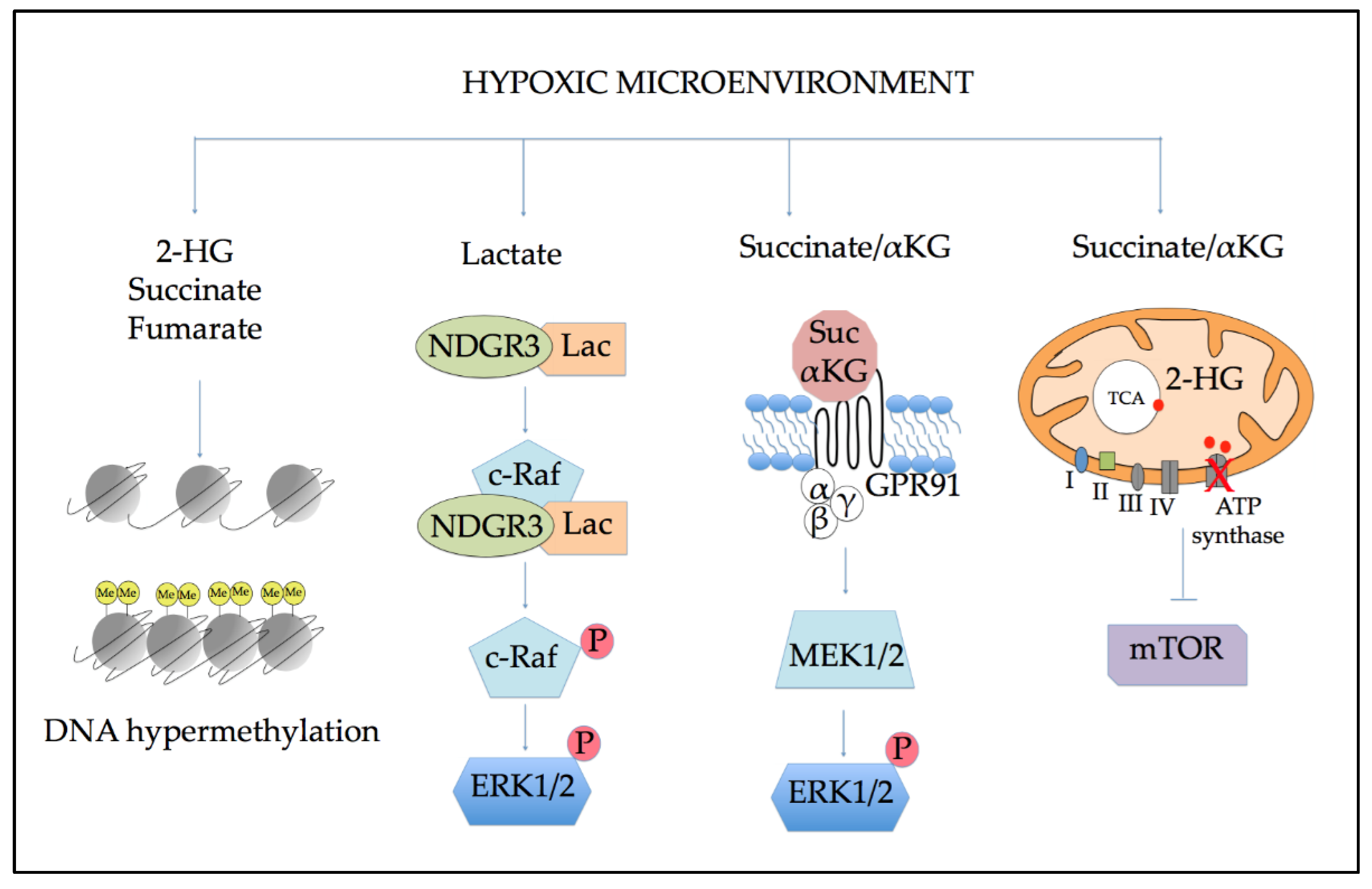
5.1. Effects of Altered Metabolism on Epigenetic Regulation
5.2. Role of Altered Metabolites in Cellular Signaling
6. Gene Expression Regulation Mediated by HIFs to Sustain AML
7. Clinical Assessment and Prognostic Value of Hypoxia
8. Therapeutic Opportunities: Targeting Hypoxia in AML
8.1. Targeting HIF Downstream Genes
8.2. Targeting Altered Metabolism
8.3. Hypoxia-Activated Prodrugs (HAP)
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| BMM | bone marrow microenvironment |
| LSCs | leukemic stem cells |
| HIF | hypoxia inducible factor |
| BM | bone marrow |
| HSCs | hematopoietic stem cells |
| PHD | prolyl hydroxylases |
| VHL | von Hippel-Lindau |
| CBP | CREB-binding protein |
| HRE | hypoxia responsive elements |
| Treg | regulatory T cells |
| STAT5 | signal transducer and activator of transcription 5 |
| VEGF | vascular endothelial growth factor |
| SDF1 | stromal cell-derived factor 1 |
| SCF | stem cell factor |
| Ang2 | angiopoietin 2 |
| Angptl | angiopoietin-likes |
| IGF-2 | insulin-like growth factor 2 |
| IGFBP | insulin-like growth factor-binding protein |
| FOXO | forkhead box O |
| CDKN | cyclin-dependent kinase inhibitor |
| ROS | reactive oxygen species |
| OXPHOS | oxidative phosphorylation |
| CML | chronic myeloid leukemia |
| TCA | tricarboxylic acid |
| UDP-GlcNAc | uridine diphosphate N-acetylglucosamine |
| UDP-GalNAc | uridine diphosphate N-acetylgalactosamine |
| 2-HG | 2-hydroxyglutarate |
| PDK1 | pyruvate dehydrogenase 1 |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| COX 4-1 | cytochrome C oxidase subunit 4 isoform 1 |
| COX 4-2 | cytochrome C oxidase subunit 4 isoform 2 |
| WHO | world health organization |
| OS | overall survival |
| EFS | event free survival |
| IDH | isocitrate dehydrogenase |
| α-KG | α-ketoglutarate |
| TET | ten-eleven translocation |
| SDH | succinate dehydrogenase |
| UQ | ubiquinone |
| UQH2 | ubiquinol |
| NDRG3 | N-Myc downstream-regulated 3 |
| ERK1/2 | extracellular-signal-regulated kinase 1/2 |
| GPR91 | G-protein-coupled receptor |
| mTOR | mammalian target of rapamycin |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| FLT3 | FMS-like tyrosine kinase 3 |
| MIF | macrophage migration factor |
| IL-8 | interleukin-8 |
| MSC | mesenchymal stromal cells |
| PI3K | phosphoinositide 3-kinase |
| PKB or AKT | protein kinase B |
| AXL | receptor tyrosine kinase |
| ITD | internal tandem duplication |
| NPM1 | nucleophosmin |
| LDH | lactate dehydrogenase |
| Ara-C | cytarabine arabinoside |
| XIAP | X-linked inhibitor of apoptosis |
| PML | promyelocytic leukemia |
| RAR- α | retinoic acid receptor-α |
| APL | acute promyelocytic leukemia |
| ATRA | trans retinoic acid |
| HAP | hypoxia-activated prodrugs |
| BCL-2 | B-cell lymphoma 2 |
| MCL-1 | myeloid cell lymphoma 1 |
| PD-L1 | programmed death ligand 1 |
| mAb | monoclonal antibody |
| MCT | monocarboxylate transporter |
| AKR1C3 | aldo-keto reductase 1C3 |
| Br-iPM | bromo-isophosphoramide mustard |
References
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Vosberg, S.; Greif, P.A. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosom. Cancer 2019, 58, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; Furudate, K.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, D.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafer, D.; Grant, S. Update on rational targeted therapy in AML. Blood Rev. 2016, 30, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef]
- Löwenberg, B.; Ossenkoppele, G.J.; Van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Maertens, J.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. High-Dose Daunorubicin in Older Patients with Acute Myeloid Leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, H.F.; Sun, Z.; Yao, X.; Litzow, M.R.; Luger, S.M.; Paietta, E.M.; Racevskis, J.; Dewald, G.W.; Ketterling, R.P.; Bennett, J.M.; et al. Anthracycline dose intensification in acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Stark, M.; Ofran, Y.; Assaraf, Y.G. Deciphering molecular mechanisms underlying chemoresistance in relapsed AML patients: Towards precision medicine overcoming drug resistance. Cancer Cell Int. 2021, 21, 53. [Google Scholar] [CrossRef]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévesque, J.-P.; Helwani, F.M.; Winkler, I.G. The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia 2010, 24, 1979–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimura, R. The significance and application of vascular niche in the development and maintenance of hematopoietic stem cells. Int. J. Hematol. 2018, 107, 642–645. [Google Scholar] [CrossRef] [Green Version]
- Eliasson, P.; Jönsson, J.-I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J. Cell. Physiol. 2010, 222, 17–22. [Google Scholar] [CrossRef]
- El-Showk, S. Low-oxygen landscape mapped in bone marrow. Nat. Middle East 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nombela-Arrieta, C.; Silberstein, L.E. The science behind the hypoxic niche of hematopoietic stem and progenitors. Hematol. Am. Soc. Hematol. Educ. Progr. 2014, 2014, 542–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef]
- Kubota, Y.; Takubo, K.; Suda, T. Bone marrow long label-retaining cells reside in the sinusoidal hypoxic niche. Biochem. Biophys. Res. Commun. 2008, 366, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Lo Celso, C.; Fleming, H.E.; Wu, J.W.; Zhao, C.X.; Miake-Lye, S.; Fujisaki, J.; Côté, D.; Rowe, D.W.; Lin, C.P.; Scadden, D.T. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 2009, 457, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Mauch, P.; Vergilio, J.-A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Umikawa, M.; Zhang, S.; Huynh, H.; Silvany, R.; Chen, B.P.C.; Chen, L.; Zhang, C.C. Ex vivo expanded hematopoietic stem cells overcome the MHC barrier in allogeneic transplantation. Cell Stem Cell 2011, 9, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Shima, H.; Takubo, K.; Iwasaki, H.; Yoshihara, H.; Gomei, Y.; Hosokawa, K.; Arai, F.; Takahashi, T.; Suda, T. Reconstitution activity of hypoxic cultured human cord blood CD34-positive cells in NOG mice. Biochem. Biophys. Res. Commun. 2009, 378, 467–472. [Google Scholar] [CrossRef]
- Shima, H.; Takubo, K.; Tago, N.; Iwasaki, H.; Arai, F.; Takahashi, T.; Suda, T. Acquisition of G0 state by CD34-positive cord blood cells after bone marrow transplantation. Exp. Hematol. 2010, 38, 1231–1240. [Google Scholar] [CrossRef]
- Moirangthem, R.D.; Singh, S.; Adsul, A.; Jalnapurkar, S.; Limaye, L.; Kale, V.P. Hypoxic Niche-Mediated Regeneration of Hematopoiesis in the Engraftment Window Is Dominantly Affected by Oxygen Tension in the Milieu. Stem Cells Dev. 2015, 24, 2423–2436. [Google Scholar] [CrossRef] [Green Version]
- Bapat, A.; Schippel, N.; Shi, X.; Jasbi, P.; Gu, H.; Kala, M.; Sertil, A.; Sharma, S. Hypoxia promotes erythroid differentiation through the development of progenitors and proerythroblasts. Exp. Hematol. 2021, 97, 32–46.e35. [Google Scholar] [CrossRef]
- Mostafa, S.S.; Miller, W.M.; Papoutsakis, E.T. Oxygen tension influences the differentiation, maturation and apoptosis of human megakaryocytes. Br. J. Haematol. 2000, 111, 879–889. [Google Scholar]
- Chen, S.; Su, Y.; Wang, J. ROS-mediated platelet generation: A microenvironment-dependent manner for megakaryocyte proliferation, differentiation, and maturation. Cell Death Dis. 2013, 4, e722. [Google Scholar] [CrossRef] [Green Version]
- Taneja, R.; Rameshwar, P.; Upperman, J.; Wang, M.T.; Livingston, D.H. Effects of hypoxia on granulocytic-monocytic progenitors in rats. Role of bone marrow stroma. Am. J. Hematol. 2000, 64, 20–25. [Google Scholar] [CrossRef]
- Zhou, H.-S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, S.M.; Gleadle, J.M.; Pugh, C.W.; Hankinson, O.; Ratcliffe, P.J. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J. Biol. Chem. 1996, 271, 15117–15123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Winkler, I.G.; Barbier, V.; Wadley, R.; Zannettino, A.C.W.; Williams, S.; Lévesque, J. Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: Serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood 2010, 116, 375–385. [Google Scholar] [CrossRef]
- Acar, M.; Kocherlakota, K.S.; Murphy, M.M.; Peyer, J.G.; Oguro, H.; Inra, C.N.; Jaiyeola, C.; Zhao, Z.; Luby-Phelps, K.; Morrison, S.J. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 2015, 526, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Yin, T.; Wiegraebe, W.; He, X.C.; Miller, D.; Stark, D.; Perko, K.; Alexander, R.; Schwartz, J.; Grindley, J.C.; et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature 2009, 457, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Guezguez, B.; Campbell, C.J.V.; Boyd, A.L.; Karanu, F.; Casado, F.L.; Di Cresce, C.; Collins, T.J.; Shapovalova, Z.; Xenocostas, A.; Bhatia, M. Regional Localization within the Bone Marrow Influences the Functional Capacity of Human HSCs. Cell Stem Cell 2013, 13, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Lo Celso, C.; Tsuyuzaki, H.; et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011, 474, 216–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehade, H.; Acolty, V.; Moser, M.; Oldenhove, G. Cutting Edge: Hypoxia-Inducible Factor 1 Negatively Regulates Th1 Function. J. Immunol. 2015, 195, 1372–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; DeBerardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Fatrai, S.; Wierenga, A.T.J.; Daenen, S.M.G.J.; Vellenga, E.; Schuringa, J.J. Identification of HIF2α as an important STAT5 target gene in human hematopoietic stem cells. Blood 2011, 117, 3320–3330. [Google Scholar] [CrossRef]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Forristal, C.E.; Nowlan, B.; Jacobsen, R.; Barbier, V.; Walkinshaw, G.; Walkley, C.; Winkler, I.; Levesque, J.P. HIF-1α is required for hematopoietic stem cell mobilization and 4-prolyl hydroxylase inhibitors enhance mobilization by stabilizing HIF-1α. Leukemia 2015, 29, 1366–1378. [Google Scholar] [CrossRef] [Green Version]
- Nombela-Arrieta, C.; Pivarnik, G.; Winkel, B.; Canty, K.J.; Harley, B.; Mahoney, J.E.; Park, S.-Y.; Lu, J.; Protopopov, A.; Silberstein, L.E. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat. Cell Biol. 2013, 15, 533–543. [Google Scholar] [CrossRef]
- Eliasson, P.; Rehn, M.; Hammar, P.; Larsson, P.; Sirenko, O.; Flippin, L.A.; Cammenga, J.; Jönsson, J.-I. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term–reconstituting hematopoietic stem cells during in vitro culture. Exp. Hematol. 2010, 38, 301–310.e2. [Google Scholar] [CrossRef] [Green Version]
- Forristal, C.E.; Winkler, I.G.; Nowlan, B.; Barbier, V.; Walkinshaw, G.; Levesque, J.-P. Pharmacologic stabilization of HIF-1α increases hematopoietic stem cell quiescence in vivo and accelerates blood recovery after severe irradiation. Blood 2013, 121, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krock, B.L.; Eisinger-Mathason, T.S.; Giannoukos, D.N.; Shay, J.E.; Gohil, M.; Lee, D.S.; Nakazawa, M.S.; Sesen, J.; Skuli, N.; Simon, M.C. The aryl hydrocarbon receptor nuclear translocator is an essential regulator of murine hematopoietic stem cell viability. Blood 2015, 125, 3263–3272. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wong, C.C.L.; Wei, H.; Gilkes, D.M.; Korangath, P.; Chaturvedi, P.; Schito, L.; Chen, J.; Krishnamachary, B.; Winnard, P.T.; et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012, 31, 1757–1770. [Google Scholar] [CrossRef]
- Kocabas, F.; Xie, L.; Xie, J.; Yu, Z.; DeBerardinis, R.J.; Kimura, W.; Thet, S.; Elshamy, A.F.; Abouellail, H.; Muralidhar, S.; et al. Hypoxic metabolism in human hematopoietic stem cells. Cell Biosci. 2015, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Miharada, K.; Karlsson, G.; Rehn, M.; Rörby, E.; Siva, K.; Cammenga, J.; Karlsson, S. Cripto Regulates Hematopoietic Stem Cells as a Hypoxic-Niche-Related Factor through Cell Surface Receptor GRP78. Cell Stem Cell 2011, 9, 330–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitart, A.V.; Subramani, C.; Armesilla-Diaz, A.; Smith, G.; Sepulveda, C.; Gezer, D.; Vukovic, M.; Dunn, K.; Pollard, P.; Holyoake, T.L.; et al. Hif-2α is not essential for cell-autonomous hematopoietic stem cell maintenance. Blood 2013, 122, 1741–1745. [Google Scholar] [CrossRef]
- Rouault-Pierre, K.; Lopez-Onieva, L.; Foster, K.; Anjos-Afonso, F.; Lamrissi-Garcia, I.; Serrano-Sanchez, M.; Mitter, R.; Ivanovic, Z.; De Verneuil, H.; Gribben, J.; et al. HIF-2α Protects Human Hematopoietic Stem/Progenitors and Acute Myeloid Leukemic Cells from Apoptosis Induced by Endoplasmic Reticulum Stress. Cell Stem Cell 2013, 13, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Naveiras, O.; Daley, G.Q. Stem cells and their niche: A matter of fate. Cell. Mol. Life Sci. 2006, 63, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, A.L.; Campbell, C.J.V.; Hopkins, C.I.; Fiebig-Comyn, A.; Russell, J.; Ulemek, J.; Foley, R.; Leber, B.; Xenocostas, A.; Collins, T.J.; et al. Niche displacement of human leukemic stem cells uniquely allows their competitive replacement with healthy HSPCs. J. Exp. Med. 2014, 211, 1925–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeb, G.; Vaughan, M.M.; McInnis, I.; Ford, L.; Ann Sait, S.N.J.; Starostik, P.; Wetzler, M.; Mashtare, T.; Wang, E.S. Hypoxia-inducible factor-1α protein expression is associated with poor survival in normal karyotype adult acute myeloid leukemia. Leuk. Res. 2011, 35, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Malek, S.N.; Zheng, P.; Liu, Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011, 8, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Vukovic, M.; Guitart, A.V.; Sepulveda, C.; Villacreces, A.; O’Duibhir, E.; Panagopoulou, T.I.; Ivens, A.; Menendez-Gonzalez, J.; Iglesias, J.M.; Allen, L.; et al. Hif-1α and Hif-2α synergize to suppress AML development but are dispensable for disease maintenance. J. Exp. Med. 2015, 212, 2223–2234. [Google Scholar] [CrossRef]
- Herst, P.M.; Howman, R.A.; Neeson, P.J.; Berridge, M.V.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J. Leukoc. Biol. 2011, 89, 51–55. [Google Scholar] [CrossRef]
- Lodi, A.; Tiziani, S.; Khanim, F.L.; Drayson, M.T.; Günther, U.L.; Bunce, C.M.; Viant, M.R. Hypoxia triggers major metabolic changes in AML cells without altering indomethacin-induced TCA cycle deregulation. ACS Chem. Biol. 2011, 6, 169–175. [Google Scholar] [CrossRef]
- Martinez, M.R.; Dias, T.B.; Natov, P.S.; Zachara, N.E. Stress-induced O-GlcNAcylation: An adaptive process of injured cells. Biochem. Soc. Trans. 2017, 45, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Pazzaglia, M.; Cerchione, C.; Soverini, S.; Cavo, M.; Montanaro, L.; Simonetti, G.; Martinelli, G. Abstract 2651: Deep hypoxia and the genomic background cooperate to shape the metabolic profile of acute myeloid leukemia cells. In Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar] [CrossRef]
- Goto, M.; Miwa, H.; Suganuma, K.; Tsunekawa-Imai, N.; Shikami, M.; Mizutani, M.; Mizuno, S.; Hanamura, I.; Nitta, M. Adaptation of leukemia cells to hypoxic condition through switching the energy metabolism or avoiding the oxidative stress. BMC Cancer 2014, 14, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, I.; Kohno, B. 18 FDG-PET/CT: 21st century approach to leukemic tumors in 124 cases. Am. J. Hematol. 2016, 91, 379–384. [Google Scholar] [CrossRef]
- Chen, W.-L.; Wang, J.; Zhao, A.; Xu, X.; Wang, Y.; Chen, T.; Li, J.; Mi, J.; Zhu, Y.; Liu, Y.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Bledea, R.; Vasudevaraja, V.; Patel, S.; Stafford, J.; Serrano, J.; Esposito, G.; Tredwin, L.M.; Goodman, N.; Kloetgen, A.; Golfinos, J.G.; et al. Functional and topographic effects on DNA methylation in IDH1/2 mutant cancers. Sci. Rep. 2019, 9, 16830. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-P.; Li, J.-T.; Qu, J.; Yin, M.; Lei, Q.-Y. Metabolite sensing and signaling in cancer. J. Biol. Chem. 2020, 295, 11938–11946. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.S.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitson, K.S.; Liénard, B.M.R.; McDonough, M.A.; Clifton, I.J.; Butler, D.; Soares, A.S.; Oldham, N.J.; McNeill, L.A.; Schofield, C.J. Structural and Mechanistic Studies on the Inhibition of the Hypoxia-inducible Transcription Factor Hydroxylases by Tricarboxylic Acid Cycle Intermediates. J. Biol. Chem. 2007, 282, 3293–3301. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.C.; Sohn, H.A.; Park, Z.; Oh, S.; Kang, Y.K.; Lee, K.; Kang, M.; Jang, Y.J.; Yang, S.; Hong, Y.K.; et al. A Lactate-Induced Response to Hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Park, K.C.; Lee, D.C.; Yeom, Y.I. NDRG3-mediated lactate signaling in hypoxia. BMB Rep. 2015, 48, 301–302. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef]
- Hakak, Y.; Lehmann-Bruinsma, K.; Phillips, S.; Le, T.; Liaw, C.; Connolly, D.T.; Behan, D.P. The role of the GPR91 ligand succinate in hematopoiesis. J. Leukoc. Biol. 2009, 85, 837–843. [Google Scholar] [CrossRef]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Padró, T.; Ruiz, S.; Bieker, R.; Bürger, H.; Steins, M.; Kienast, J.; Büchner, T.; Berdel, W.E.; Mesters, R.M. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood 2000, 95, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Hussong, J.W.; Rodgers, G.M.; Shami, P.J. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood 2000, 95, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.; Hattori, K.; Zhu, Z.; Heissig, B.; Choy, M.; Lane, W.; Wu, Y.; Chadburn, A.; Hyjek, E.; Gill, M.; et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J. Clin. Investig. 2000, 106, 511–521. [Google Scholar] [CrossRef]
- Fiegl, M.; Samudio, I.; Clise-Dwyer, K.; Burks, J.K.; Mnjoyan, Z.; Andreeff, M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood 2009, 113, 1504–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sison, E.A.R.; McIntyre, E.; Magoon, D.; Brown, P. Dynamic Chemotherapy-Induced Upregulation of CXCR4 Expression: A Mechanism of Therapeutic Resistance in Pediatric AML. Mol. Cancer Res. 2013, 11, 1004–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konoplev, S.; Rassidakis, G.Z.; Estey, E.; Kantarjian, H.; Liakou, C.I.; Huang, X.; Xiao, L.; Andreeff, M.; Konopleva, M.; Medeiros, L.J. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer 2007, 109, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, E.J.C.; Pavic, B.; Löwenberg, B.; Ploemacher, R.E. Relation between CXCR-4 expression, FLT3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood 2004, 104, 550–557. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Shafat, M.S.; Sun, Y.; Marlein, C.R.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Zhou, Z.; Collins, A.; Bowles, K.M.; et al. HIF1α drives chemokine factor pro-tumoral signaling pathways in acute myeloid leukemia. Oncogene 2018, 37, 2676–2686. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Shafat, M.S.; Sun, Y.; Marlein, C.R.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Zhou, Z.; Collins, A.; Bowles, K.M.; et al. MIF-Induced Stromal PKCβ/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017, 77, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Kuett, A.; Rieger, C.; Perathoner, D.; Herold, T.; Wagner, M.; Sironi, S.; Sotlar, K.; Horny, H.; Deniffel, C.; Drolle, H.; et al. IL-8 as mediator in the microenvironment-leukaemia network in acute myeloid leukaemia. Sci. Rep. 2015, 5, 18411. [Google Scholar] [CrossRef]
- Sironi, S.; Wagner, M.; Kuett, A.; Drolle, H.; Polzer, H.; Spiekermann, K.; Rieger, C.; Fiegl, M. Microenvironmental hypoxia regulates FLT3 expression and biology in AML. Sci. Rep. 2015, 5, 17550. [Google Scholar] [CrossRef]
- Dumas, P.-Y.; Naudin, C.; Martin-Lannerée, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossée, M.; Giese, A.; et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef] [Green Version]
- Elhoseiny, S.M. Hypoxia-Inducible Factor 1 Alpha (HIF-1α) and Its Prognostic Value in Acute Myeloid Leukemia. Hematol. Transfus. Int. J. 2017, 4, 19–25. [Google Scholar] [CrossRef]
- Geva, M.; Shouval, R.; Fein, J.A.; Danylesko, I.; Shem-Tov, N.; Yerushalmi, R.; Shimoni, A.; Nagler, A. Lactate Dehydrogenase Is a Key Prognostic Factor in Acute Myeloid Leukemia and Lymphoma Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2019, 134, 3304. [Google Scholar] [CrossRef]
- Drolle, H.; Wagner, M.; Vasold, J.; Kütt, A.; Deniffel, C.; Sotlar, K.; Sironi, S.; Herold, T.; Rieger, C.; Fiegl, M. Hypoxia regulates proliferation of acute myeloid leukemia and sensitivity against chemotherapy. Leuk. Res. 2015, 39, 779–785. [Google Scholar] [CrossRef]
- Matsunaga, T.; Imataki, O.; Torii, E.; Kameda, T.; Shide, K.; Shimoda, H.; Kamiunten, A.; Sekine, M.; Taniguchi, Y.; Yamamoto, S.; et al. Elevated HIF-1α expression of acute myelogenous leukemia stem cells in the endosteal hypoxic zone may be a cause of minimal residual disease in bone marrow after chemotherapy. Leuk. Res. 2012, 36, e122–e124. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of Hypoxia-inducible Factor-1α Protein in Hypoxia Occurs Independently of Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- Coltella, N.; Percio, S.; Valsecchi, R.; Cuttano, R.; Guarnerio, J.; Ponzoni, M.; Pandolfi, P.P.; Melillo, G.; Pattini, L.; Bernardi, R. HIF factors cooperate with PML-RARα to promote acute promyelocytic leukemia progression and relapse. EMBO Mol. Med. 2014, 6, 640–650. [Google Scholar] [CrossRef]
- Abraham, M.; Klein, S.; Bulvik, B.; Wald, H.; Weiss, I.D.; Olam, D.; Weiss, L.; Beider, K.; Eizenberg, O.; Wald, O.; et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia 2017, 31, 2336–2346. [Google Scholar] [CrossRef]
- Borthakur, G.; Ofran, Y.; Tallman, M.S.; Foran, J.; Uy, G.L.; DiPersio, J.F.; Showel, M.M.; Shimoni, A.; Nagler, A.; Rowe, J.M.; et al. BL-8040 CXCR4 antagonist is safe and demonstrates antileukemic activity in combination with cytarabine for the treatment of relapsed/refractory acute myelogenous leukemia: An open-label safety and efficacy phase 2a study. Cancer 2021, 127, 1246–1259. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The Plasma Membrane Lactate Transporter MCT4, but Not MCT1, Is Up-regulated by Hypoxia through a HIF-1α-dependent Mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Saulle, E.; Spinello, I.; Quaranta, M.T.; Pasquini, L.; Pelosi, E.; Iorio, E.; Castelli, G.; Chirico, M.; Pisanu, M.E.; Ottone, T.; et al. Targeting Lactate Metabolism by Inhibiting MCT1 or MCT4 Impairs Leukemic Cell Proliferation, Induces Two Different Related Death-Pathways and Increases Chemotherapeutic Sensitivity of Acute Myeloid Leukemia Cells. Front. Oncol. 2021, 10, 3394. [Google Scholar] [CrossRef]
- Singleton, R.S.; Guise, C.P.; Ferry, D.M.; Pullen, S.M.; Dorie, M.J.; Brown, J.M.; Patterson, A.V.; Wilson, W.R. DNA Cross-Links in Human Tumor Cells Exposed to the Prodrug PR-104A: Relationships to Hypoxia, Bioreductive Metabolism, and Cytotoxicity. Cancer Res. 2009, 69, 3884–3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, A.V.; Ferry, D.M.; Edmunds, S.J.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; et al. Mechanism of Action and Preclinical Antitumor Activity of the Novel Hypoxia-Activated DNA Cross-Linking Agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guise, C.P.; Abbattista, M.R.; Singleton, R.S.; Holford, S.D.; Connolly, J.; Dachs, G.U.; Fox, S.B.; Pollock, R.; Harvey, J.; Guilford, P.; et al. The Bioreductive Prodrug PR-104A Is Activated under Aerobic Conditions by Human Aldo-Keto Reductase 1C3. Cancer Res. 2010, 70, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, S.M.F.; Gu, Y.; Manesh, D.M.; El-Hoss, J.; Jing, D.; MacKenzie, K.L.; Guise, C.P.; Foehrenbacher, A.; Pullen, S.M.; Benito, J.; et al. A novel fluorometric assay for aldo-keto reductase 1C3 predicts metabolic activation of the nitrogen mustard prodrug PR-104A in human leukaemia cells. Biochem. Pharmacol. 2014, 88, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Thall, P.F.; Yi, C.A.; Borthakur, G.; Coveler, A.; Bueso-Ramos, C.; Benito, J.; Konoplev, S.; Gu, Y.; Ravandi, F.; et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica 2015, 100, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Portwood, S.; Lal, D.; Hsu, Y.; Vargas, R.; Johnson, M.K.; Wetzler, M.; Hart, C.P.; Wang, E.S. Activity of the Hypoxia-Activated Prodrug, TH-302, in Preclinical Human Acute Myeloid Leukemia Models. Clin. Cancer Res. 2013, 19, 6506–6519. [Google Scholar] [CrossRef] [Green Version]
- Benito, J.; Ramirez, M.S.; Millward, N.Z.; Velez, J.; Harutyunyan, K.G.; Lu, H.; Shi, Y.; Matre, P.; Jacamo, R.; Ma, H.; et al. Hypoxia-Activated Prodrug TH-302 Targets Hypoxic Bone Marrow Niches in Preclinical Leukemia Models. Clin. Cancer Res. 2016, 22, 1687–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badar, T.; Handisides, D.R.; Benito, J.M.; Richie, M.A.; Borthakur, G.; Jabbour, E.; Harutyunyan, K.; Konoplev, S.; Faderl, S.; Kroll, S.; et al. Phase I study of evofosfamide, an investigational hypoxia-activated prodrug, in patients with advanced leukemia. Am. J. Hematol. 2016, 91, 800–805. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanism of Action | Clinical Trial ID | References Preclinical Studies |
|---|---|---|---|
| BL8040 | CXCR4 inhibition | NCT01838395 NCT03154827 | [99] |
| IMC-1C11 | VEGFR-2 inhibition | na | [82] |
| IACS-010759 | Complex I (OXPHOS) inhibition | NCT02882321 | [101] |
| AR-C155858/Syrosingopine | MCT1 and MCT4 inhibition | na | [103] |
| PR104 | DNA cross-links | NCT01037556 | [104,105,108] |
| TH-302 | DNA cross-links | NCT01149915 | [109,110,111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, S.; Mancini, M.; De Santis, S.; Monaldi, C.; Cavo, M.; Soverini, S. The Role of Hypoxic Bone Marrow Microenvironment in Acute Myeloid Leukemia and Future Therapeutic Opportunities. Int. J. Mol. Sci. 2021, 22, 6857. https://doi.org/10.3390/ijms22136857
Bruno S, Mancini M, De Santis S, Monaldi C, Cavo M, Soverini S. The Role of Hypoxic Bone Marrow Microenvironment in Acute Myeloid Leukemia and Future Therapeutic Opportunities. International Journal of Molecular Sciences. 2021; 22(13):6857. https://doi.org/10.3390/ijms22136857
Chicago/Turabian StyleBruno, Samantha, Manuela Mancini, Sara De Santis, Cecilia Monaldi, Michele Cavo, and Simona Soverini. 2021. "The Role of Hypoxic Bone Marrow Microenvironment in Acute Myeloid Leukemia and Future Therapeutic Opportunities" International Journal of Molecular Sciences 22, no. 13: 6857. https://doi.org/10.3390/ijms22136857
APA StyleBruno, S., Mancini, M., De Santis, S., Monaldi, C., Cavo, M., & Soverini, S. (2021). The Role of Hypoxic Bone Marrow Microenvironment in Acute Myeloid Leukemia and Future Therapeutic Opportunities. International Journal of Molecular Sciences, 22(13), 6857. https://doi.org/10.3390/ijms22136857