
VAV Proteins as Double Agents in Cancer: Oncogenes with Tumor Suppressor Roles



{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
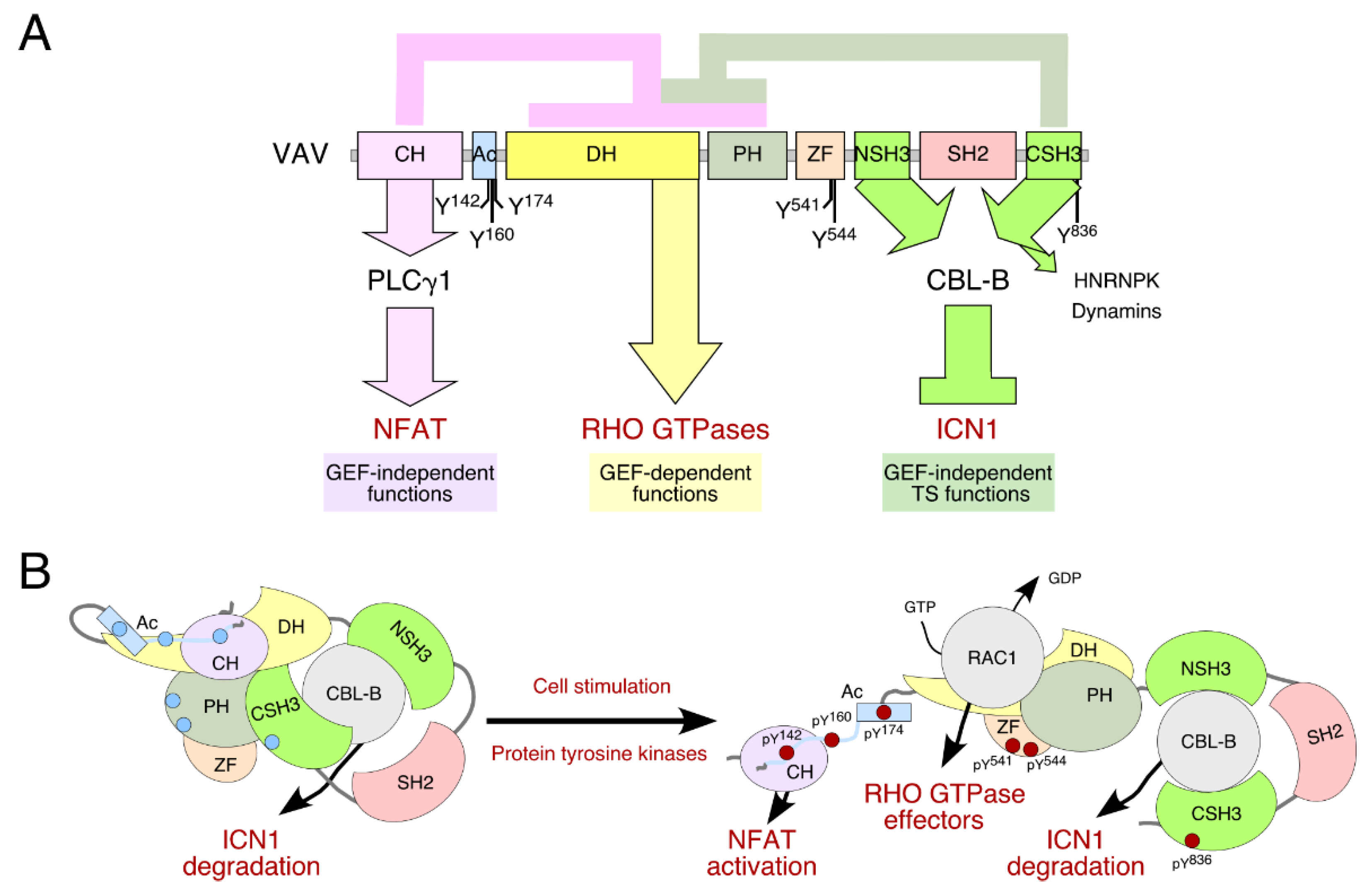
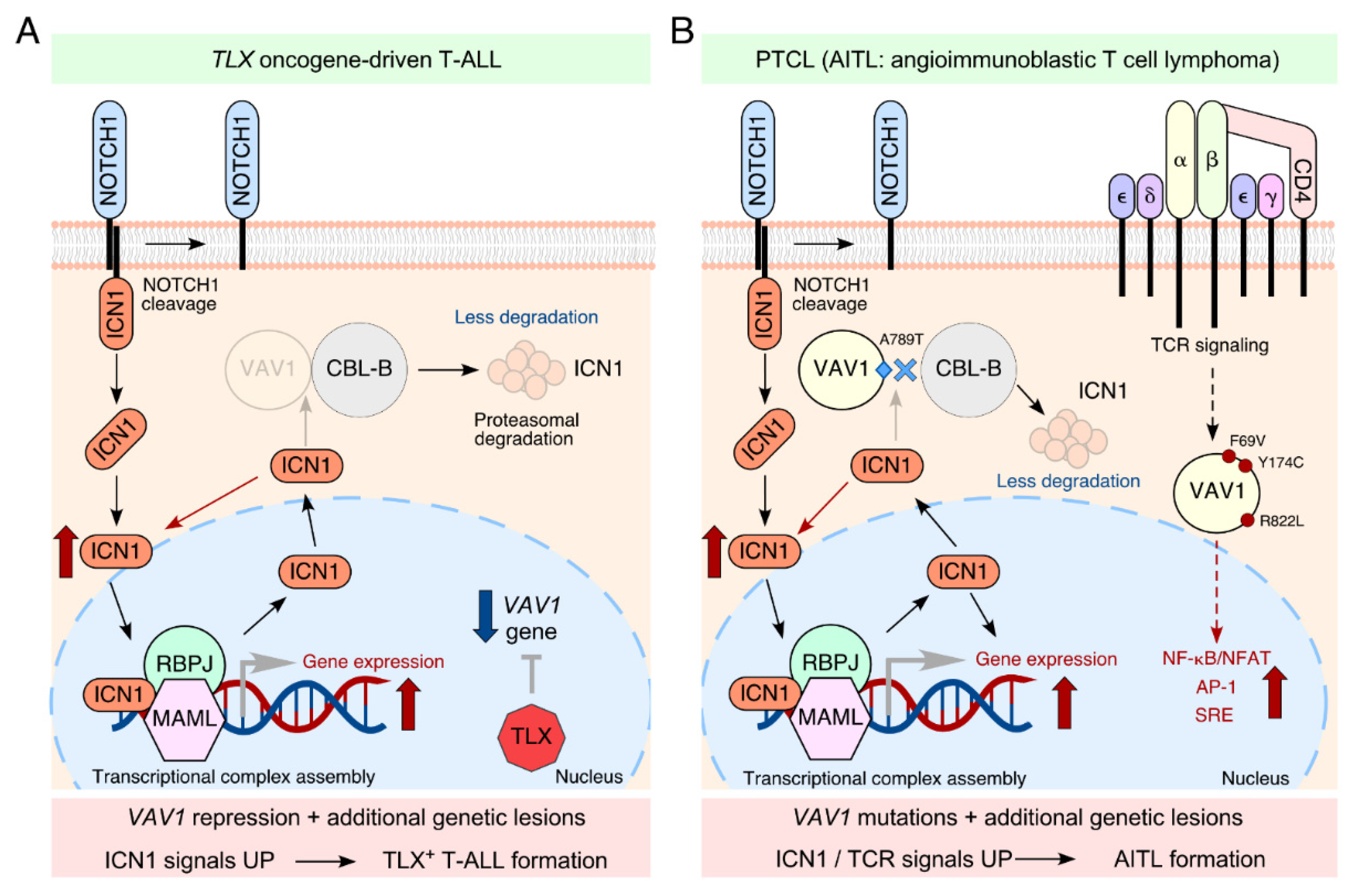
2. VAV1 as a Tumor Suppressor in T-ALLs
3. How Widespread Is This Tumor Suppressor Function in Neoplastic Processes?
4. VAV2 and VAV3 in Cancer: Could They Also Be Involved in Tumor Suppressor Pathways?
5. Final Remarks
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Marshall, C.J. RHO-GTPases and cancer. Nat. Rev. Cancer 2002, 2, 133–142. [Google Scholar] [CrossRef]
- Robles-Valero, J.; Lorenzo-Martín, L.F.; Fernández-Pisonero, I.; Bustelo, X.R. Rho guanosine nucleotide exchange factors are not such bad guys after all in cancer. Small GTPases 2020, 11, 233–239. [Google Scholar] [CrossRef]
- Bustelo, X.R. Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 2014, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Fdez, S.; Bustelo, X.R. The Vav GEF Family: An Evolutionary and Functional Perspective. Cells 2019, 8, 465. [Google Scholar] [CrossRef] [Green Version]
- Crespo, P.; Schuebel, K.E.; Ostrom, A.A.; Gutkind, J.S.; Bustelo, X.R. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature 1997, 385, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Couceiro, J.R.; Martín-Bermudo, M.D.; Bustelo, X.R. Phylogenetic conservation of the regulatory and functional properties of the Vav oncoprotein family. Exp. Cell Res. 2005, 308, 364–380. [Google Scholar] [CrossRef] [Green Version]
- Bustelo, X.R. Vav proteins, adaptors and cell signaling. Oncogene 2001, 20, 6372–6381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citterio, C.; Menacho-Márquez, M.; García-Escudero, R.; Larive, R.M.; Barreiro, O.; Sánchez-Madrid, F.; Paramio, J.M.; Bustelo, X.R. The rho exchange factors vav2 and vav3 control a lung metastasis-specific transcriptional program in breast cancer cells. Sci. Signal. 2012, 5, ra71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menacho-Márquez, M.; García-Escudero, R.; Ojeda, V.; Abad, A.; Delgado, P.; Costa, C.; Ruiz, S.; Alarcón, B.; Paramio, J.M.; Bustelo, X.R. The Rho exchange factors Vav2 and Vav3 favor skin tumor initiation and promotion by engaging extracellular signaling loops. PLoS Biol. 2013, 11, e1001615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles-Valero, J.; Lorenzo-Martín, L.F.; Menacho-Márquez, M.; Fernández-Pisonero, I.; Abad, A.; Camós, M.; Toribio, M.L.; Espinosa, L.; Bigas, A.; Bustelo, X.R. A Paradoxical Tumor-Suppressor Role for the Rac1 Exchange Factor Vav1 in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2017, 32, 608–623.e609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzav, S.; Martin-Zanca, D.; Barbacid, M. vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. EMBO J. 1989, 8, 2283–2290. [Google Scholar] [CrossRef]
- Chang, K.H.; Sanchez-Aguilera, A.; Shen, S.; Sengupta, A.; Madhu, M.N.; Ficker, A.M.; Dunn, S.K.; Kuenzi, A.M.; Arnett, J.L.; Santho, R.A.; et al. Vav3 collaborates with p190-BCR-ABL in lymphoid progenitor leukemogenesis, proliferation, and survival. Blood 2012, 120, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Abate, F.; da Silva-Almeida, A.C.; Zairis, S.; Robles-Valero, J.; Couronne, L.; Khiabanian, H.; Quinn, S.A.; Kim, M.Y.; Laginestra, M.A.; Kim, C.; et al. Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T-cell lymphomas. Proc. Natl. Acad. Sci. USA 2017, 114, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo’, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [Green Version]
- Boddicker, R.L.; Razidlo, G.L.; Dasari, S.; Zeng, Y.; Hu, G.; Knudson, R.A.; Greipp, P.T.; Davila, J.I.; Johnson, S.H.; Porcher, J.C.; et al. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood 2016, 128, 1234–1245. [Google Scholar] [CrossRef]
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia 2018, 32, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Park, J.; Yang, J.; Wenzel, A.T.; Ramachandran, A.; Lee, W.J.; Daniels, J.C.; Kim, J.; Martinez-Escala, E.; Amankulor, N.; Pro, B.; et al. Genomic analysis of 220 CTCLs identifies a novel recurrent gain-of-function alteration in RLTPR (p.Q575E). Blood 2017, 130, 1430–1440. [Google Scholar] [CrossRef]
- Vallois, D.; Dobay, M.P.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.E.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef]
- Ruiz, S.; Santos, E.; Bustelo, X.R. The use of knockout mice reveals a synergistic role of the Vav1 and Rasgrf2 gene deficiencies in lymphomagenesis and metastasis. PLoS ONE 2009, 4, e8229. [Google Scholar] [CrossRef] [Green Version]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikawa, K.; Miletic, A.V.; Alt, F.W.; Faccio, R.; Brown, T.; Hoog, J.; Fredericks, J.; Nishi, S.; Mildiner, S.; Moores, S.L.; et al. Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J. Exp. Med. 2003, 198, 1595–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.P.; Pieters, R.; et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustelo, X.R.; Crespo, P.; Lopez-Barahona, M.; Gutkind, J.S.; Barbacid, M. Cbl-b, a member of the Sli-1/c-Cbl protein family, inhibits Vav-mediated c-Jun N-terminal kinase activation. Oncogene 1997, 15, 2511–2520. [Google Scholar] [CrossRef] [Green Version]
- Della Gatta, G.; Palomero, T.; Perez-Garcia, A.; Ambesi-Impiombato, A.; Bansal, M.; Carpenter, Z.W.; De Keersmaecker, K.; Sole, X.; Xu, L.; Paietta, E.; et al. Reverse engineering of TLX oncogenic transcriptional networks identifies RUNX1 as tumor suppressor in T-ALL. Nat. Med. 2012, 18, 436–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voena, C.; Chiarle, R. RHO Family GTPases in the Biology of Lymphoma. Cells 2019, 8, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Zapico, M.E.; Gonzalez-Paz, N.C.; Weiss, E.; Savoy, D.N.; Molina, J.R.; Fonseca, R.; Smyrk, T.C.; Chari, S.T.; Urrutia, R.; Billadeau, D.D. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell 2005, 7, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazer, G.; Idelchuk, Y.; Schapira, V.; Pikarsky, E.; Katzav, S. The haematopoietic specific signal transducer Vav1 is aberrantly expressed in lung cancer and plays a role in tumourigenesis. J. Pathol. 2009, 219, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Barreira, M.; Fabbiano, S.; Couceiro, J.R.; Torreira, E.; Martinez-Torrecuadrada, J.L.; Montoya, G.; Llorca, O.; Bustelo, X.R. The C-Terminal SH3 Domain Contributes to the Intramolecular Inhibition of Vav Family Proteins. Sci. Signal. 2014, 7, ra35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, S.; Li, J.; Yang, H.; Zeng, H.; Han, W.; Zhang, J.; Lee, M.; Moczygemba, M.; Isgandarova, S.; Yang, Y.; et al. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J. Clin. Investig. 2017, 127, 2998–3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, K.; Sakata-Yanagimoto, M.; Fujisawa, M.; Sakamoto, T.; Miyoshi, H.; Suehara, Y.; Nguyen, T.B.; Suma, S.; Yanagimoto, S.; Shiraishi, Y.; et al. VAV1 mutations contribute to development of T-cell neoplasms in mice. Blood 2020, 136, 3018–3032. [Google Scholar] [CrossRef]
- Fiore, D.; Cappelli, L.V.; Broccoli, A.; Zinzani, P.L.; Chan, W.C.; Inghirami, G. Peripheral T cell lymphomas: From the bench to the clinic. Nat. Rev. Cancer 2020, 20, 323–342. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Suen, K.L.; Michael, W.M.; Dreyfuss, G.; Barbacid, M. Association of the vav proto-oncogene product with poly(rC)-specific RNA-binding proteins. Mol. Cell Biol. 1995, 15, 1324–1332. [Google Scholar] [CrossRef] [Green Version]
- Gomez, T.S.; Hamann, M.J.; McCarney, S.; Savoy, D.N.; Lubking, C.M.; Heldebrant, M.P.; Labno, C.M.; McKean, D.J.; McNiven, M.A.; Burkhardt, J.K.; et al. Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat. Immunol. 2005, 6, 261–270. [Google Scholar] [CrossRef]
- Gallardo, M.; Lee, H.J.; Zhang, X.; Bueso-Ramos, C.; Pageon, L.R.; McArthur, M.; Multani, A.; Nazha, A.; Manshouri, T.; Parker-Thornburg, J.; et al. hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies. Cancer Cell 2015, 28, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, C.; Guo, W.; Zheng, S.; Sun, Z.; Geng, X. DNM3 Attenuates Hepatocellular Carcinoma Growth by Activating P53. Med. Sci. Monit. 2016, 22, 197–205. [Google Scholar] [CrossRef]
- Houlard, M.; Arudchandran, R.; Regnier-Ricard, F.; Germani, A.; Gisselbrecht, S.; Blank, U.; Rivera, J.; Varin-Blank, N. Vav1 is a component of transcriptionally active complexes. J. Exp. Med. 2002, 195, 1115–1127. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Zhang, N.; Wan, Y.; Liu, Y.; Zhang, L.; Pan, S.; Zhang, C.; Zhang, H.; Cao, Y. The interaction between Vav1 and EBNA1 promotes survival of Burkitt’s lymphoma cells by down-regulating the expression of Bim. Biochem. Biophys. Res. Commun. 2019, 511, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Lyons, L.S.; Fahrenholtz, C.D.; Wu, F.; Farooq, A.; Balkan, W.; Burnstein, K.L. A novel nuclear role for the Vav3 nucleotide exchange factor in androgen receptor coactivation in prostate cancer. Oncogene 2012, 31, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo-Martín, L.F.; Rodríguez-Fdez, S.; Fabbiano, S.; Abad, A.; García-Macías, M.C.; Dosil, M.; Cuadrado, M.; Robles-Valero, J.; Bustelo, X.R. Vav2 pharmaco-mimetic mice reveal the therapeutic value and caveats of the catalytic inactivation of a Rho exchange factor. Oncogene 2020, 39, 5098–5111. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Martín, L.F.; Fernández-Parejo, N.; Menacho-Márquez, M.; Rodríguez-Fdez, S.; Robles-Valero, J.; Zumalave, S.; Fabbiano, S.; Pascual, G.; García-Pedrero, J.M.; Abad, A.; et al. VAV2 signaling promotes regenerative proliferation in both cutaneous and head and neck squamous cell carcinoma. Nat. Commun. 2020, 11, 4788. [Google Scholar] [CrossRef]
- Lorenzo-Martín, L.F.; Citterio, C.; Menacho-Márquez, M.; Conde, J.; Larive, R.M.; Rodríguez-Fdez, S.; García-Escudero, R.; Robles-Valero, J.; Cuadrado, M.; Fernández-Pisonero, I.; et al. Vav proteins maintain epithelial traits in breast cancer cells using miR-200c-dependent and independent mechanisms. Oncogene 2019, 38, 209–227. [Google Scholar] [CrossRef]
- Cortes, J.R.; Ambesi-Impiombato, A.; Couronné, L.; Quinn, S.A.; Kim, C.S.; da Silva Almeida, A.C.; West, Z.; Belver, L.; Martin, M.S.; Scourzic, L.; et al. RHOA G17V Induces T Follicular Helper Cell Specification and Promotes Lymphomagenesis. Cancer Cell 2018, 33, 259–273.e257. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.Y.; Brown, L.; Stevenson, K.; deSouza, T.; Aster, J.C.; Louissaint, A.; Weinstock, D.M. RhoA G17V is sufficient to induce autoimmunity and promotes T-cell lymphomagenesis in mice. Blood 2018, 132, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Demeulemeester, J.; Wedge, D.C.; Vollan, H.K.M.; Pitt, J.J.; Russnes, H.G.; Pandey, B.P.; Nilsen, G.; Nord, S.; Bignell, G.R.; et al. Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors. Nat. Commun. 2017, 8, 1221. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Diamantopoulou, Z.; White, G.; Fadlullah, M.Z.H.; Dreger, M.; Pickering, K.; Maltas, J.; Ashton, G.; MacLeod, R.; Baillie, G.S.; Kouskoff, V.; et al. TIAM1 Antagonizes TAZ/YAP Both in the Destruction Complex in the Cytoplasm and in the Nucleus to Inhibit Invasion of Intestinal Epithelial Cells. Cancer Cell 2017, 31, 621–634.e626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elly, C.; Witte, S.; Zhang, Z.; Rosnet, O.; Lipkowitz, S.; Altman, A.; Liu, Y.C. Tyrosine phosphorylation and complex formation of Cbl-b upon T cell receptor stimulation. Oncogene 1999, 18, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Armato, M.A.; Yankee, T.M. Immature single-positive CD8+ thymocytes represent the transition from Notch-dependent to Notch-independent T-cell development. Int. Immunol. 2011, 23, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Vaqué, J.P.; Gómez-López, G.; Monsálvez, V.; Varela, I.; Martínez, N.; Pérez, C.; Domínguez, O.; Graña, O.; Rodríguez-Peralto, J.L.; Rodríguez-Pinilla, S.M.; et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014, 123, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010. [Google Scholar] [CrossRef] [Green Version]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuadrado, M.; Robles-Valero, J. VAV Proteins as Double Agents in Cancer: Oncogenes with Tumor Suppressor Roles. Biology 2021, 10, 888. https://doi.org/10.3390/biology10090888
Cuadrado M, Robles-Valero J. VAV Proteins as Double Agents in Cancer: Oncogenes with Tumor Suppressor Roles. Biology. 2021; 10(9):888. https://doi.org/10.3390/biology10090888
Chicago/Turabian StyleCuadrado, Myriam, and Javier Robles-Valero. 2021. "VAV Proteins as Double Agents in Cancer: Oncogenes with Tumor Suppressor Roles" Biology 10, no. 9: 888. https://doi.org/10.3390/biology10090888
APA StyleCuadrado, M., & Robles-Valero, J. (2021). VAV Proteins as Double Agents in Cancer: Oncogenes with Tumor Suppressor Roles. Biology, 10(9), 888. https://doi.org/10.3390/biology10090888