
Cross Talk of Macrophages with Tumor Microenvironment Cells and Modulation of Macrophages in Cancer by Virotherapy




Abstract
:1. Introduction
2. Macrophage–Cancer Cell Cross Talk and TME Remodeling
2.1. Cancer Cell Signals Reprogram Macrophage Function and Polarization
2.2. Macrophage Signals to Cancer Cells
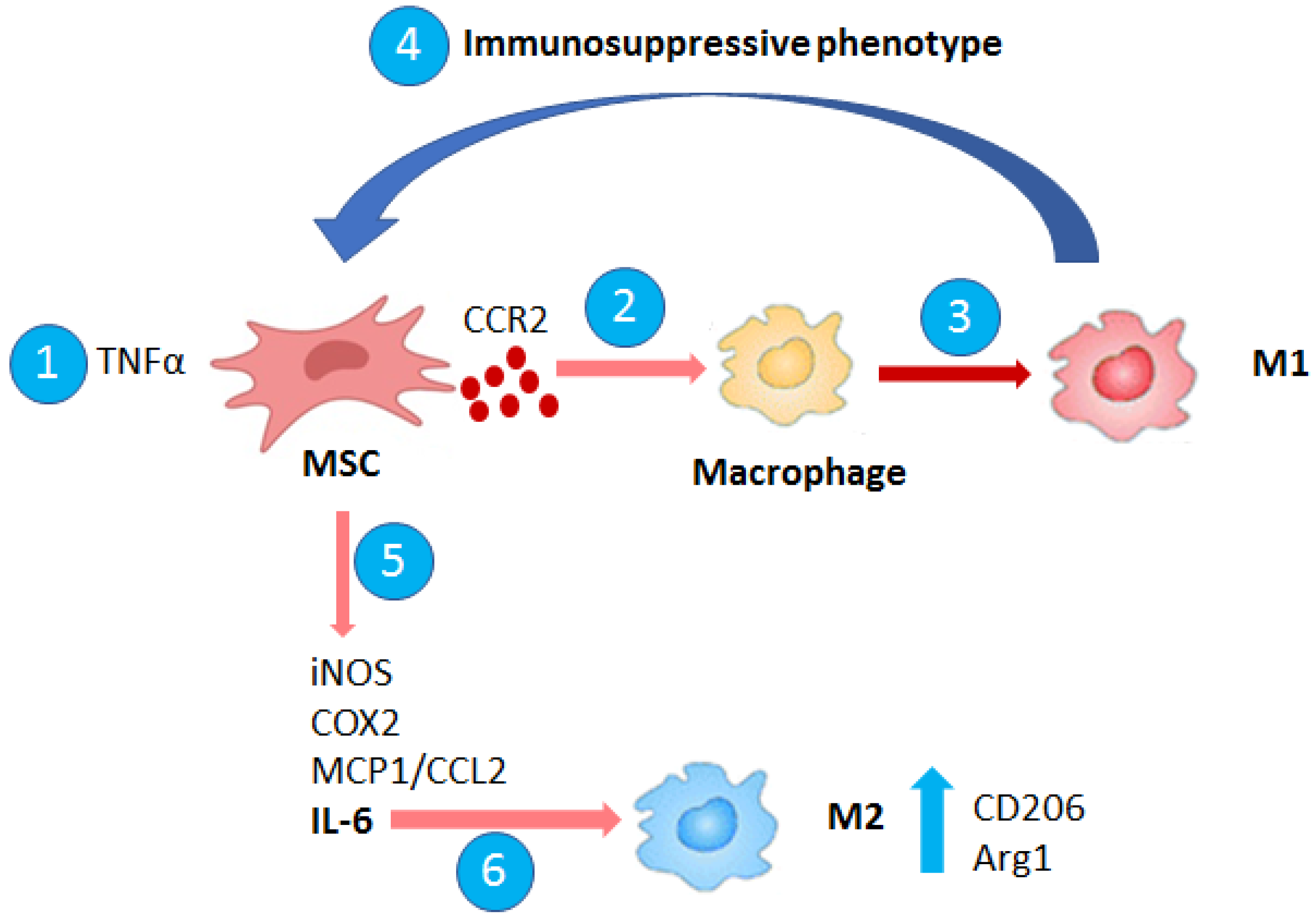
3. Macrophage–MSC Cross Talk in Cancer and TME Remodeling
Macrophage–MSC Interaction
4. Macrophage–Cancer-Associated Fibroblast (CAF) Cross Talk and TME Remodeling
Macrophage–CAF Interaction

{kind=link}
| Type of Tumor | Macrophage/CAF Interaction | Ref. |
|---|---|---|
| Prostate cancer |
| [30,36] |
| Breast cancer |
| [34] |
| Neuroblastoma |
| [35] |
| Human colorectal cancer |
| [37] |
| Hepatocellular carcinoma |
| [38] |
5. Macrophages and Virotherapy
5.1. Macrophages in Tumor Response to OVs
5.2. Modulation of Macrophage Phenotype by OVs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Halldén, G.; Portella, G. Oncolytic virotherapy with modified adenoviruses and novel therapeutic targets. Expert Opin. Ther. Targets 2012, 16, 945–958. [Google Scholar] [CrossRef]
- Di Somma, S.; Iannuzzi, C.A.; Passaro, C.; Forte, I.M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; et al. The Oncolytic Virus dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front. Oncol. 2019, 9, 564. [Google Scholar] [CrossRef] [Green Version]
- Malfitano, A.M.; di Somma, S.; Iannuzzi, C.A.; Pentimalli, F.; Portella, G. Virotherapy: From single agents to combinatorial treatments. Biochem. Pharmacol. 2020, 177, 113986. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Somma, S.D.; Prevete, N.; Portella, G. Virotherapy as a Potential Therapeutic Approach for the Treatment of Aggressive Thyroid Cancer. Cancers 2019, 11, 1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannuzzi, C.A.; Indovina, P.; Forte, I.M.; di Somma, S.; Malfitano, A.M.; Bruno, M.; Portella, G.; Pentimalli, F.; Giordano, A. Pharmacological Inhibition of WEE1 Potentiates the Antitumoral Effect of the dl922-947 Oncolytic Virus in Malignant Mesothelioma Cell Lines. Int. J. Mol. Sci. 2020, 21, 7333. [Google Scholar] [CrossRef]
- Passaro, C.; Volpe, M.; Botta, G.; Scamardella, E.; Perruolo, G.; Gillespie, D.; Libertini, S.; Portella, G. PARP inhibitor olaparib increases the oncolytic activity of dl922-947 in in vitro and in vivo model of anaplastic thyroid carcinoma. Mol. Oncol. 2015, 9, 78–92. [Google Scholar] [CrossRef]
- Hashimoto, O.; Yoshida, M.; Koma, Y.; Yanai, T.; Hasegawa, D.; Kosaka, Y.; Nishimura, N.; Yokozaki, H. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J. Pathol. 2016, 240, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Pisanti, S.; Napolitano, F.; di Somma, S.; Martinelli, R.; Portella, G. Tumor-Associated Macrophage Status in Cancer Treatment. Cancers 2020, 12, 1987. [Google Scholar] [CrossRef]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Cervantes-Villagrana, R.D.; Albores-García, D.; Cervantes-Villagrana, A.R.; García-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Targeted Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Di Somma, S.; Amato, J.; Iaccarino, N.; Pagano, B.; Randazzo, A.; Portella, G.; Malfitano, A.M. G-Quadruplex Binders Induce Immunogenic Cell Death Markers in Aggressive Breast Cancer Cells. Cancers 2019, 11, 1797. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Sun, L.; Lyu, X.; Ai, X.; Du, D.; Su, N.; Li, H.; Zhang, L.; Yu, J.; Yuan, S. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: A positive metabolic feedback loop. Oncotarget 2017, 8, 110426–110443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.; Kim, S.; Hong, B.J.; Lee, C.J.; Kim, Y.E.; Bok, S.; Oh, J.M.; Gwak, S.H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019, 79, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, H.; Zhou, Q.; Zheng, S.; Li, G.; Lin, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef] [Green Version]
- Castro-Manrreza, M.E. Participation of mesenchymal stem cells in the regulation of immune response and cancer development. Bol. Med. Hosp. Infant. Mex. 2016, 73, 380–387. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Kansy, B.A.; Dißmann, P.A.; Hemeda, H.; Bruderek, K.; Westerkamp, A.M.; Jagalski, V.; Schuler, P.; Kansy, K.; Lang, S.; Dumitru, C.A.; et al. The bidirectional tumor—Mesenchymal stromal cell interaction promotes the progression of head and neck cancer. Stem Cell Res. Ther. 2014, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, E.; Brannon, A.L.; del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katanov, C.; Lerrer, S.; Liubomirski, Y.; Leider-Trejo, L.; Meshel, T.; Bar, J.; Feniger-Barish, R.; Kamer, I.; Soria-Artzi, G.; Kahani, H.; et al. Regulation of the inflammatory profile of stromal cells in human breast cancer: Prominent roles for TNF-α and the NF-κB pathway. Stem Cell Res. Ther. 2015, 6, 87. [Google Scholar] [CrossRef] [Green Version]
- Melissari, M.T.; Chalkidi, N.; Sarris, M.E.; Koliaraki, V. Fibroblast Reprogramming in Gastrointestinal Cancer. Front. Cell Dev. Biol. 2020, 8, 630. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Trivanovic, D.; Jaukovic, A.; Santibanez, J.F.; Bugarski, D. Metabolic Plasticity of Stem Cells and Macrophages in Cancer. Front. Immunol. 2017, 8, 939. [Google Scholar] [CrossRef] [Green Version]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippman, M.E.; Datar, R.H.; et al. Identification of Cancer-Associated Fibroblasts in Circulating Blood from Patients with Metastatic Breast Cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Takeya, M. CAFs and TAMs: Maestros of the tumour microenvironment. J. Pathol. 2017, 241, 313–315. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chai, S.; Wang, W.; Wan, C.; Zhang, F.; Li, Y.; Wang, F. Macrophages activate mesenchymal stem cells to acquire cancer-associated fibroblast-like features resulting in gastric epithelial cell lesions and malignant transformation in vitro. Oncol. Lett. 2019, 17, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Sakakura, K.; Kawabata-Iwakawa, R.; Rokudai, S.; Toyoda, M.; Nishiyama, M.; Chikamatsu, K. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol. Immunother. 2015, 64, 1407–1417. [Google Scholar] [CrossRef]
- Gok Yavuz, B.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1(+) TAMs. Sci. Rep. 2019, 9, 3172. [Google Scholar] [CrossRef] [PubMed]
- Larsson, K.; Kock, A.; Idborg, H.; Arsenian Henriksson, M.; Martinsson, T.; Johnsen, J.I.; Korotkova, M.; Kogner, P.; Jakobsson, P.J. COX/mPGES-1/PGE2 pathway depicts an inflammatory-dependent high-risk neuroblastoma subset. Proc. Natl. Acad. Sci. USA 2015, 112, 8070–8075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.R.; Slavin, S.; Da, J.; Hsu, I.; Luo, J.; Xiao, G.Q.; Ding, J.; Chou, F.J.; Yeh, S. Estrogen receptor α in cancer associated fibroblasts suppresses prostate cancer invasion via reducing CCL5, IL6 and macrophage infiltration in the tumor microenvironment. Mol. Cancer 2016, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Morine, Y.; Tokuda, K.; Yamada, S.; Saito, Y.; Nishi, M.; Ikemoto, T.; Shimada, M. Cancer-associated fibroblast-induced M2-polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor-1 pathway. Int. J. Oncol. 2021, 59, 59. [Google Scholar] [CrossRef] [PubMed]
- De Silva, N.; Atkins, H.; Kirn, D.H.; Bell, J.C.; Breitbach, C.J. Double trouble for tumours: Exploiting the tumour microenvironment to enhance anticancer effect of oncolytic viruses. Cytokine Growth Factor Rev. 2010, 21, 135–141. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomantonio, M.A.; Sterea, A.M.; Kim, Y.; Paulo, J.A.; Clements, D.R.; Kennedy, B.E.; Bydoun, M.J.; Shi, G.; Waisman, D.M.; Gygi, S.P.; et al. Quantitative Proteome Responses to Oncolytic Reovirus in GM-CSF- and M-CSF-Differentiated Bone Marrow-Derived Cells. J. Proteome Res. 2020, 19, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, E.; Larionova, I.; Choinzonov, E.; Kzhyshkowska, J. Monocytes and Macrophages as Viral Targets and Reservoirs. Int. J. Mol. Sci. 2018, 19, 2821. [Google Scholar] [CrossRef] [Green Version]
- Pasquereau, S.; Al Moussawi, F.; Karam, W.; Diab Assaf, M.; Kumar, A.; Herbein, G. Cytomegalovirus, Macrophages and Breast Cancer. Open Virol. J. 2017, 11, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polzin, M.; McCanless, J.; Owen, S.; Sizemore, D.; Lucero, E.; Fuller, R.; Neufeld, H.S.; Seals, D.F.; Ahmed, M. Oncolytic vesicular stomatitis viruses selectively target M2 macrophages. Virus Res. 2020, 284, 197991. [Google Scholar] [CrossRef]
- Jakeman, P.G.; Hills, T.E.; Fisher, K.D.; Seymour, L.W. Macrophages and their interactions with oncolytic viruses. Curr. Opin. Pharmacol. 2015, 24, 23–29. [Google Scholar] [CrossRef]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef] [Green Version]
- Kleijn, A.; Kloezeman, J.; Treffers-Westerlaken, E.; Fulci, G.; Leenstra, S.; Dirven, C.; Debets, R.; Lamfers, M. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PLoS ONE 2014, 9, e97495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofman, L.; Lawler, S.E.; Lamfers, M.L.M. The Multifaceted Role of Macrophages in Oncolytic Virotherapy. Viruses 2021, 13, 1570. [Google Scholar] [CrossRef]
- Fu, X.; Tao, L.; Wu, W.; Zhang, X. Arming HSV-Based Oncolytic Viruses with the Ability to Redirect the Host’s Innate Antiviral Immunity to Attack Tumor Cells. Mol. Ther. Oncolytics 2020, 19, 33–46. [Google Scholar] [CrossRef]
- Marelli, G.; Chard Dunmall, L.S.; Yuan, M.; di Gioia, C.; Miao, J.; Cheng, Z.; Zhang, Z.; Liu, P.; Ahmed, J.; Gangeswaran, R.; et al. A systemically deliverable Vaccinia virus with increased capacity for intertumoral and intratumoral spread effectively treats pancreatic cancer. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Tan, D.Q.; Zhang, L.; Ohba, K.; Ye, M.; Ichiyama, K.; Yamamoto, N. Macrophage response to oncolytic paramyxoviruses potentiates virus-mediated tumor cell killing. Eur. J. Immunol. 2016, 46, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, A.; Winder, N.; Atkinson, E.; Al-Janabi, H.; Allen, R.J.; Hughes, R.; Moamin, M.; Louie, R.; Evans, D.; Hutchinson, M.; et al. Macrophages Mediate the Antitumor Effects of the Oncolytic Virus HSV1716 in Mammary Tumors. Mol. Cancer Ther. 2021, 20, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Passaro, C.; Borriello, F.; Vastolo, V.; di Somma, S.; Scamardella, E.; Gigantino, V.; Franco, R.; Marone, G.; Portella, G. The oncolytic virus dl922-947 reduces IL-8/CXCL8 and MCP-1/CCL2 expression and impairs angiogenesis and macrophage infiltration in anaplastic thyroid carcinoma. Oncotarget 2016, 7, 1500–1515. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, W.B.L.; Kleijn, A.; Teunissen, C.E.; Voerman, J.S.A.; Teodosio, C.; Noske, D.P.; van Dongen, J.J.M.; Dirven, C.M.F.; Lamfers, M.L.M. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro-Oncology 2018, 20, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Somma, S.; Napolitano, F.; Portella, G.; Malfitano, A.M. Cross Talk of Macrophages with Tumor Microenvironment Cells and Modulation of Macrophages in Cancer by Virotherapy. Biomedicines 2021, 9, 1309. https://doi.org/10.3390/biomedicines9101309
Di Somma S, Napolitano F, Portella G, Malfitano AM. Cross Talk of Macrophages with Tumor Microenvironment Cells and Modulation of Macrophages in Cancer by Virotherapy. Biomedicines. 2021; 9(10):1309. https://doi.org/10.3390/biomedicines9101309
Chicago/Turabian StyleDi Somma, Sarah, Fabiana Napolitano, Giuseppe Portella, and Anna Maria Malfitano. 2021. "Cross Talk of Macrophages with Tumor Microenvironment Cells and Modulation of Macrophages in Cancer by Virotherapy" Biomedicines 9, no. 10: 1309. https://doi.org/10.3390/biomedicines9101309
APA StyleDi Somma, S., Napolitano, F., Portella, G., & Malfitano, A. M. (2021). Cross Talk of Macrophages with Tumor Microenvironment Cells and Modulation of Macrophages in Cancer by Virotherapy. Biomedicines, 9(10), 1309. https://doi.org/10.3390/biomedicines9101309