
In a Prediabetic Model, Empagliflozin Improves Hepatic Lipid Metabolism Independently of Obesity and before Onset of Hyperglycemia

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Characterization of Metabolic Parameters in HHTg Rats
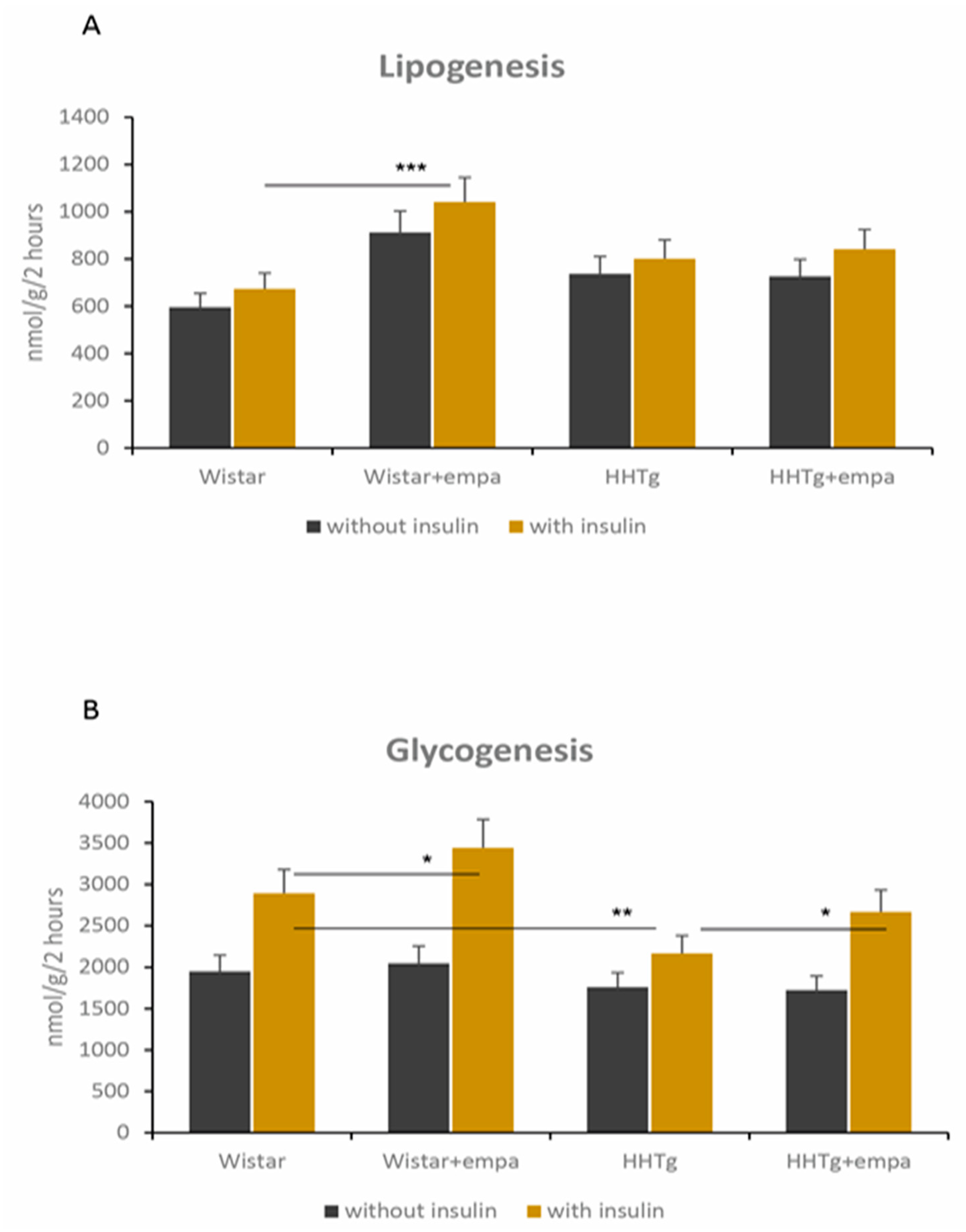
2.2. Effect of Empagliflozin Treatment on Basic Metabolic Parameters and Insulin Sensitivity
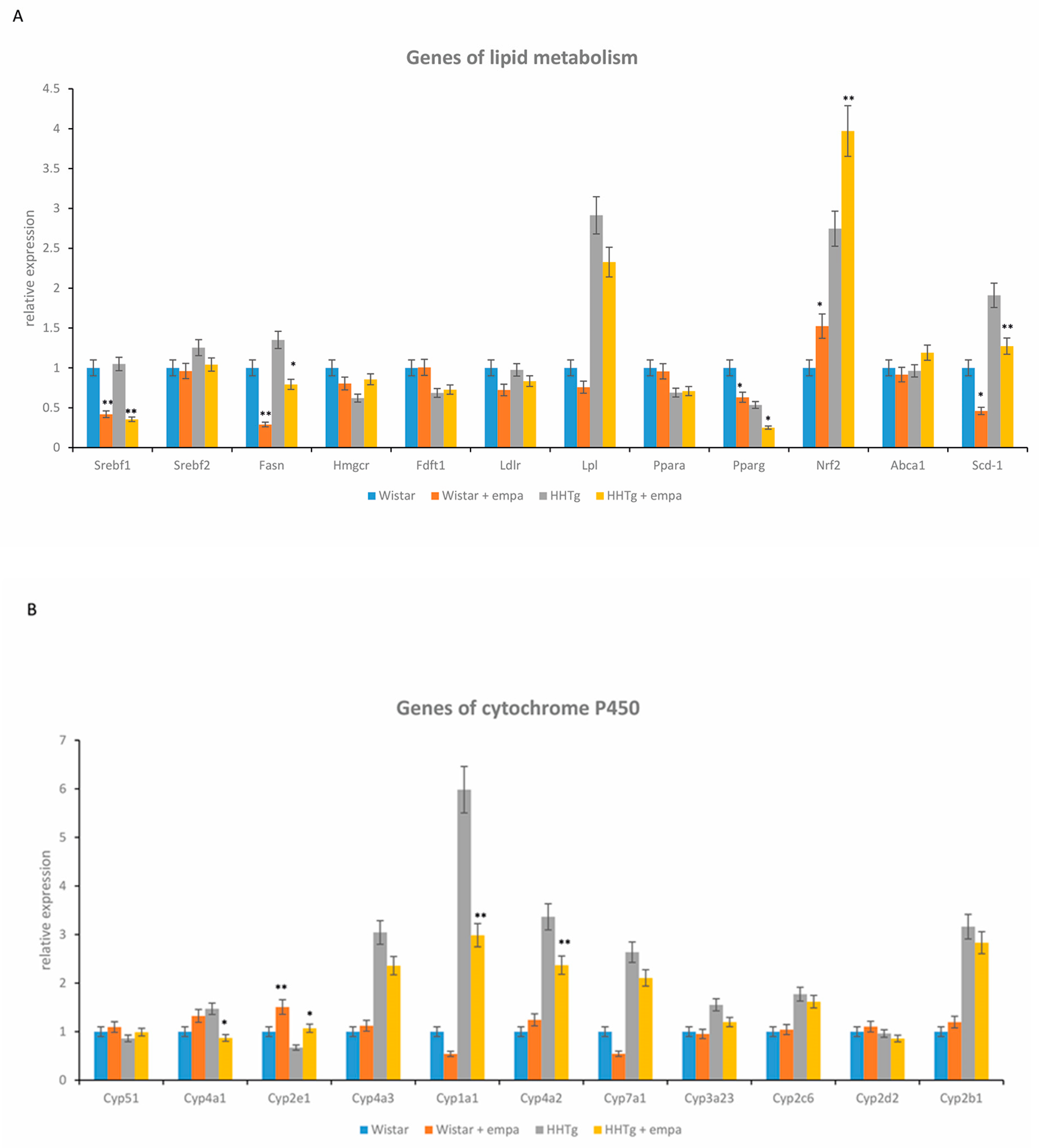
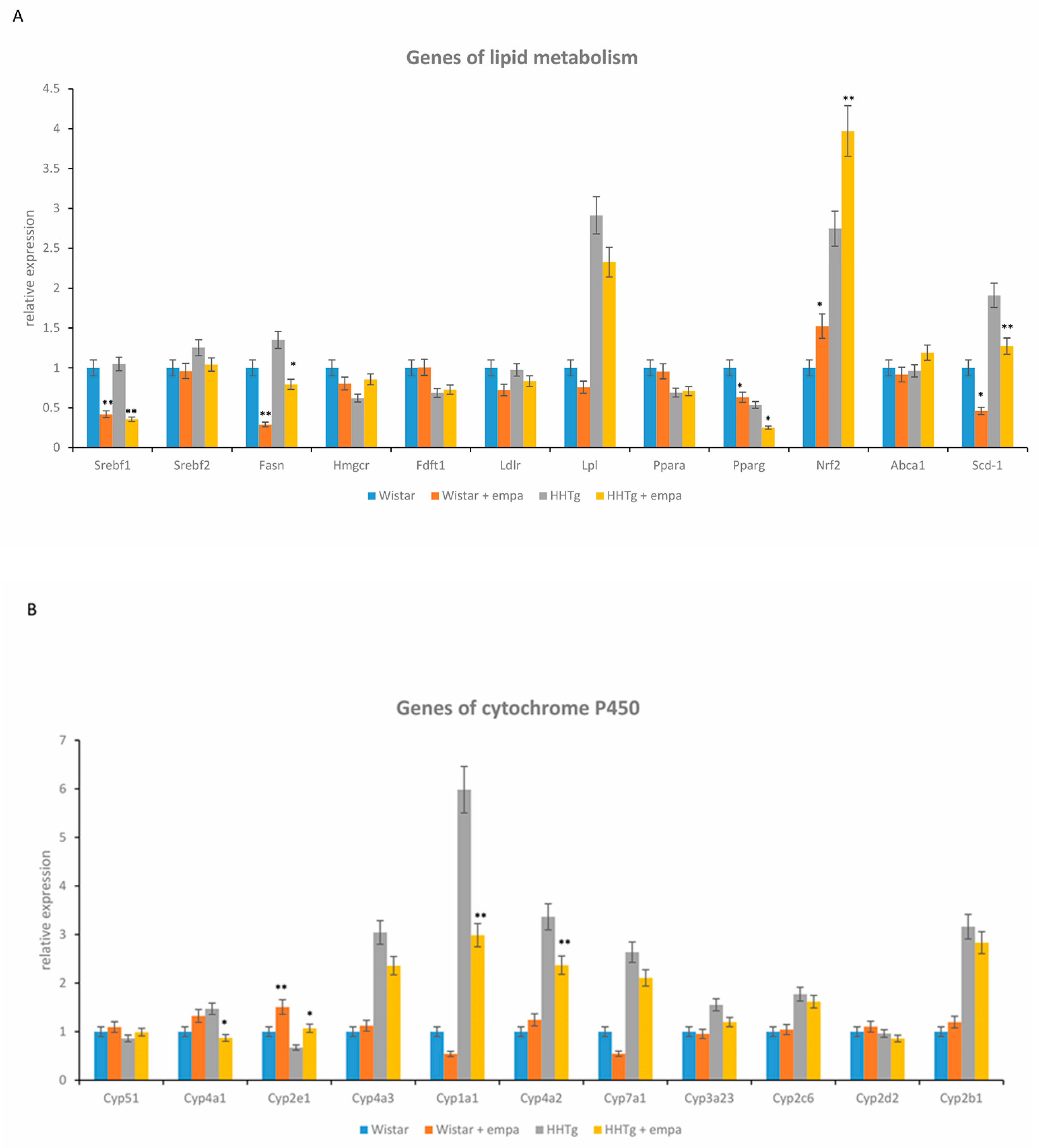
2.3. Effect of Empagliflozin Treatment on Serum Lipids and Hepatic Lipid Metabolism
2.4. Effect of Empagliflozin Treatment on Hepatic Cytochrome P450 Family Proteins
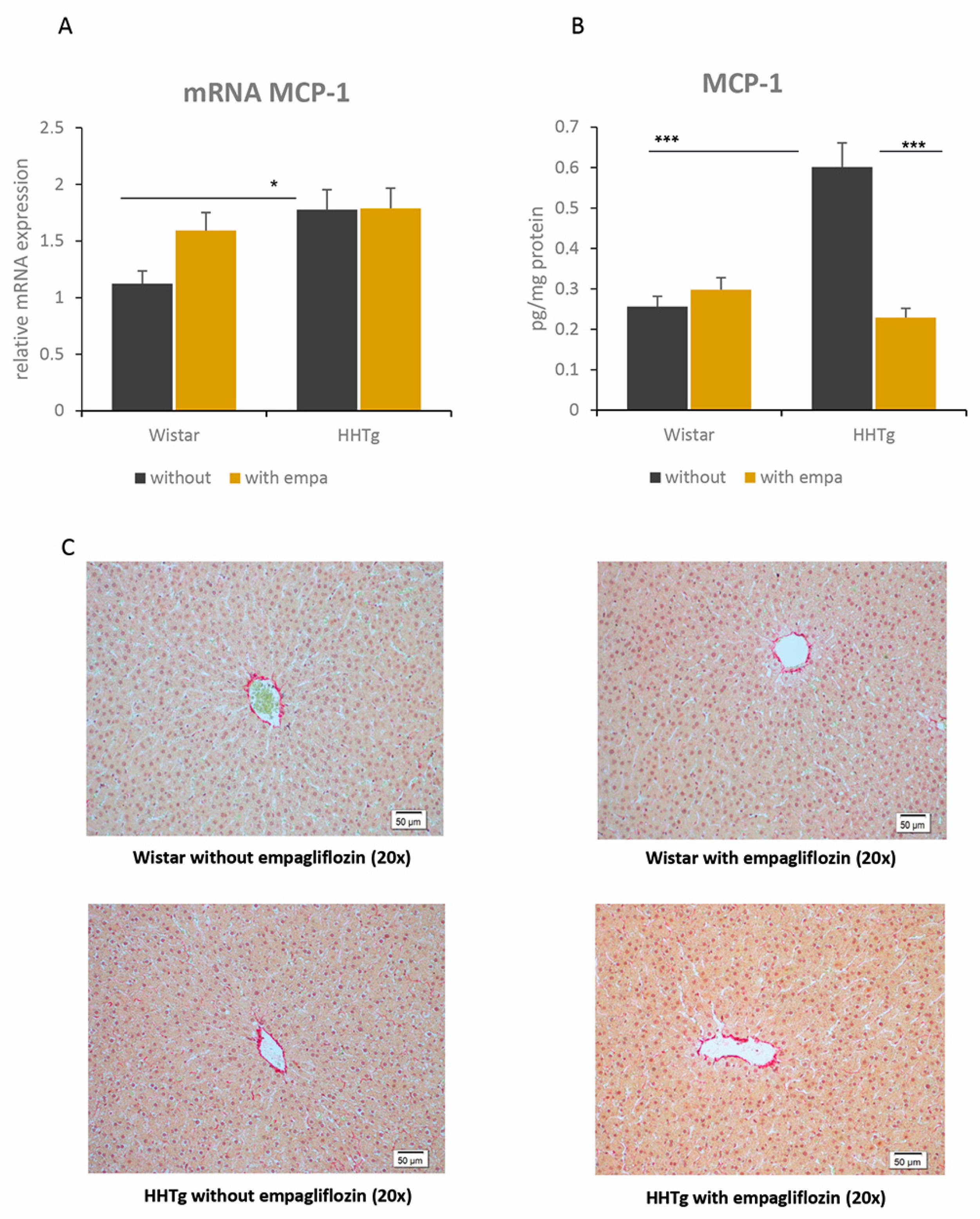
2.5. Effect of Empagliflozin Treatment on Hepatokines and Inflammatory and Oxidative Stress Parameters
3. Discussion
4. Materials and Methods
4.1. Animals and Diet
4.2. Analytic Methods, Biochemical Analysis
4.3. Oxidative Stress Parameters
4.4. Relative mRNA Expression
4.5. Histological Evaluation
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | acetyl-coenzyme A carboxylase |
| AMPK | adenosine monophosphate-activated protein kinase |
| CYP450 | cytochrome P450 |
| DAG | diacylglyceroles |
| FAS | fatty acid synthase |
| FGF21 | fibroblast growth factor 21 |
| GSH | reduced form of glutathione |
| GSSG | oxidized form of glutathione |
| HMGCR | 3-hydroxy-3-methylglutaryl-coenzyme A reductase |
| 4-HNE | 4-hydroxynonenale |
| hsCRP | high-sensitivity C-reactive protein |
| IL-6 | interleukin 6 |
| MDA | malondialdehyde |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| NAFLD | non-alcoholic fatty liver disease |
| NEFA | non-esterified fatty acid |
| NF-κB | nuclear factor kappa B |
| NRF2 | nuclear factor erythroid-2-related factor 2 |
| PPARα | peroxisome proliferator-activated receptor alpha |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| SCD | stearoyl-coenzyme A desaturase |
| SREBP1 | sterol regulatory element-binding protein 1 |
| TAG | triacylglycerols |
| TNFα | tumor necrosis factor alpha |
References
- Targher, G.; Day, C.P.; Bonora, E. Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-S.; Lee, B.-W. Beneficial effect of anti-diabetic drugs for nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2020, 26, 430–443. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Tomlinson, J. Mechanisms in endocrinology: Non-alcoholic fatty liver disease in common endocrine disorders. Eur. J. Endocrinol. 2013, 169, R27–R37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Kim, W.R. Nonobese Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.K.; Strong, J. The Pleiotropic Effects of Sodium–Glucose Cotransporter-2 Inhibitors: Beyond the Glycemic Benefit. Diabetes Ther. 2019, 10, 1771–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchay, M.S.; Farooqui, K.J.; Mishra, S.K.; Mithal, A. Glucose Lowering Efficacy and Pleiotropic Effects of Sodium-Glucose Cotransporter 2 Inhibitors. Adv. Exp. Med. Biol. 2020, 1307, 213–230. [Google Scholar] [CrossRef]
- Kahl, S.; Gancheva, S.; Straßburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2019, 43, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chehrehgosha, H.; Sohrabi, M.R.; Ismail-Beigi, F.; Malek, M.; Babaei, M.R.; Zamani, F.; Ajdarkosh, H.; Khoonsari, M.; Fallah, A.E.; Khamseh, M.E. Empagliflozin Improves Liver Steatosis and Fibrosis in Patients with Non-Alcoholic Fatty Liver Disease and Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Diabetes Ther. 2021, 12, 843–861. [Google Scholar] [CrossRef] [PubMed]
- Taheri, H.; Malek, M.; Ismail-Beigi, F.; Zamani, F.; Sohrabi, M.; Babaei, M.R.; Khamseh, M.E. Effect of Empagliflozin on Liver Steatosis and Fibrosis in Patients With Non-Alcoholic Fatty Liver Disease Without Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. Adv. Ther. 2020, 37, 4697–4708. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Petracca, G.; Csermely, A.; Beatrice, G.; Targher, G. Sodium-Glucose Cotransporter-2 Inhibitors for Treatment of Nonalcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Metabolites 2020, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE((−/−)) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. [Google Scholar] [CrossRef] [PubMed]
- Petito-Da-Silva, T.I.; Souza-Mello, V.; Barbosa-Da-Silva, S. Empaglifozin mitigates NAFLD in high-fat-fed mice by alleviating insulin resistance, lipogenesis and ER stress. Mol. Cell. Endocrinol. 2019, 498, 110539. [Google Scholar] [CrossRef] [PubMed]
- Perakakis, N.; Chrysafi, P.; Feigh, M.; Veidal, S.; Mantzoros, C. Empagliflozin Improves Metabolic and Hepatic Outcomes in a Non-Diabetic Obese Biopsy-Proven Mouse Model of Advanced NASH. Int. J. Mol. Sci. 2021, 22, 6332. [Google Scholar] [CrossRef] [PubMed]
- Zicha, J.; Pechánová, O.; Cacányiová, S.; Cebová, M.; Kristek, F.; Török, J.; Simko, F.; Dobesová, Z.; Kunes, J. Hereditary hypertriglyceridemic rat: A suitable model of cardiovascular disease and metabolic syndrome? Physiol. Res. 2006, 55 (Suppl. 1), 49–63. [Google Scholar]
- Vrána, A.; Kazdová, L. The hereditary hypertriglyceridemic nonobese rat: An experimental model of human hypertriglyceridemia. Transplant. Proc. 1990, 22, 2579. [Google Scholar]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metab. Clin. Exp. 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Schork, A.; Saynisch, J.; Vosseler, A.; Jaghutriz, B.A.; Heyne, N.; Peter, A.; Häring, H.-U.; Stefan, N.; Fritsche, A.; Artunc, F. Effect of SGLT2 inhibitors on body composition, fluid status and renin–angiotensin–aldosterone system in type 2 diabetes: A prospective study using bioimpedance spectroscopy. Cardiovasc. Diabetol. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Ferrannini, G.; Hach, T.; Crowe, S.; Sanghvi, A.; Hall, K.D.; Ferrannini, E. Energy Balance After Sodium–Glucose Cotransporter 2 Inhibition. Diabetes Care 2015, 38, 1730–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, Y.-J.; You, Y.; Moon, M.K.; Yoon, K.; Ahn, Y.; Ko, S. Effect of sodium-glucose cotransporter 2 inhibitor, empagliflozin, and α-glucosidase inhibitor, voglibose, on hepatic steatosis in an animal model of type 2 diabetes. J. Cell. Biochem. 2019, 120, 8534–8546. [Google Scholar] [CrossRef] [PubMed]
- Hojná, S.; Rauchová, H.; Malínská, H.; Marková, I.; Hüttl, M.; Papoušek, F.; Behuliak, M.; Miklánková, D.; Vaňourková, Z.; Neckář, J.; et al. Antihypertensive and metabolic effects of empagliflozin in Ren-2 transgenic rats, an experimental non-diabetic model of hypertension. Biomed. Pharmacother. 2021, 144, 112246. [Google Scholar] [CrossRef]
- Iwata, K.; Kinoshita, M.; Yamada, S.; Imamura, T.; Uenoyama, Y.; Tsukamura, H.; Maeda, K.-I. Involvement of brain ketone bodies and the noradrenergic pathway in diabetic hyperphagia in rats. J. Physiol. Sci. 2011, 61, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, S.; Gastaldelli, A. The role of the liver in the modulation of glucose and insulin in non alcoholic fatty liver disease and type 2 diabetes. Curr. Opin. Pharmacol. 2020, 55, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef]
- Pokharel, A.; Kc, S.; Thapa, P.; Karki, N.; Shrestha, R.; Jaishi, B.; Paudel, M.S. The Effect of Empagliflozin on Liver Fat in Type 2 Diabetes Mellitus Patients With Non-Alcoholic Fatty Liver Disease. Cureus 2021, 13, 16687. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.; Klöting, N.; Mark, M.; Mayoux, E.; Klein, T.; Blüher, M. The SGLT2 inhibitor empagliflozin improves insulin sensitivity in db/db mice both as monotherapy and in combination with linagliptin. Metabolism 2016, 65, 114–123. [Google Scholar] [CrossRef]
- Endo, T.; Samokhvalov, V.; Darwesh, A.M.; Khey, K.M.W.; El-Sherbeni, A.; El-Kadi, A.O.S.; Machida, T.; Hirafuji, M.; Seubert, J.M. DHA and 19,20-EDP induce lysosomal-proteolytic-dependent cytotoxicity through de novo ceramide production in H9c2 cells with a glycolytic profile. Cell Death Discov. 2018, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Uno, T.; Nakano, R.; Kitagawa, R.; Okada, M.; Kanamaru, K.; Takenaka, S.; Uno, Y.; Imaishi, H.; Kitagawa, R. Metabolism of steroids by cytochrome P450 2C9 variants. Biopharm. Drug Dispos. 2018, 39, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, M.C.; Mayoux, E.; Vallon, V. A comprehensive review of the pharmacodynamics of the SGLT2 inhibitor empagliflozin in animals and humans. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 801–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Tan, W.; Liu, X.; Deng, L.; Huang, L.; Wang, X.; Gao, X. New insight and potential therapy for NAFLD: CYP2E1 and flavonoids. Biomed. Pharmacother. 2021, 137, 111326. [Google Scholar] [CrossRef] [PubMed]
- Harjumäki, R.; Pridgeon, C.; Ingelman-Sundberg, M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.-M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.-S.; Lee, M.; Mun, S.J.; Hong, S.-H.; Lee, H.-J.; Ahn, H.-S.; Chung, K.-S.; Kim, G.-H.; Son, M.J. Targeting CYP4A attenuates hepatic steatosis in a novel multicellular organotypic liver model. J. Biol. Eng. 2019, 13, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, S.; Zhou, Y.; Su, W.; Ruan, X.; Wang, B.; Zheng, F.; Warner, M.; Gustafsson, J.-Å.; Guan, Y. Ablation of cytochrome P450 omega-hydroxylase 4A14 gene attenuates hepatic steatosis and fibrosis. Proc. Natl. Acad. Sci. USA 2017, 114, 3181–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Bao, J.; Cao, Y.-R.; Gao, H.-F.; Jin, Y. Cytochrome P450 1A1 (CYP1A1) Catalyzes Lipid Peroxidation of Oleic Acid-Induced HepG2 Cells. Biochemistry 2018, 83, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Dogru, T.; Kirik, A.; Gurel, H.; Rizvi, A.; Rizzo, M.; Sonmez, A. The Evolving Role of Fetuin-A in Nonalcoholic Fatty Liver Disease: An Overview from Liver to the Heart. Int. J. Mol. Sci. 2021, 22, 6627. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Emoto, M.; Inaba, M. Fetuin-A: A multifunctional protein. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 124–146. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Lee, W.; Jung, S.-H.; Park, J.; Sim, H.; Choi, Y.-J.; Park, Y.-J.; Chung, Y.; Lee, B.-H. Hepatic upregulation of fetuin-A mediates acetaminophen-induced liver injury through activation of TLR4 in mice. Biochem. Pharmacol. 2019, 166, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Xu, C.; Lin, J.; Li, Y. Role of hepatokines in non-alcoholic fatty liver disease. J. Transl. Intern. Med. 2019, 7, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Kong, Y.; Peng, D. Fibroblast growth factor 21 in lipid metabolism and non-alcoholic fatty liver disease. Clin. Chim. Acta 2019, 498, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Eren, F.; Yonal, O.; Kurt, R.; Aktas, B.; Celikel, C.A.; Ozdogan, O.; Imeryuz, N.; Kalayci, C.; Avsar, E. Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur. J. Clin. Investig. 2010, 40, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Ashino, T.; Ohkubo-Morita, H.; Yamamoto, M.; Yoshida, T.; Numazawa, S. Possible involvement of nuclear factor erythroid 2-related factor 2 in the gene expression of Cyp2b10 and Cyp2a5. Redox Biol. 2014, 2, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhamid, A.M.; Elsheakh, A.R.; Abdelaziz, R.R.; Suddek, G.M. Empagliflozin ameliorates ethanol-induced liver injury by modulating NF-κB/Nrf-2/PPAR-γ interplay in mice. Life Sci. 2020, 256, 117908. [Google Scholar] [CrossRef] [PubMed]
- Malinska, H.; Hüttl, M.; Oliyarnyk, O.; Bratova, M.; Kazdova, L. Conjugated linoleic acid reduces visceral and ectopic lipid accumulation and insulin resistance in chronic severe hypertriacylglycerolemia. Nutrition 2015, 31, 1045–1051. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wistar | Wistar + Empa | HHTg | HHTg + Empa | PS | PT | PI | |
|---|---|---|---|---|---|---|---|
| Body weight (g) | 568 ± 9 | 524 ± 14 ** | 465 ± 9 | 418 ± 9 ** | <0.001 | ˂0.001 | n.s. |
| Epididymal adipose tissue weight (mg/g) | 1.75 ± 0.09 | 1.44 ± 0.10 ** | 1.66 ± 0.03 | 1.40 ± 0.05 * | n.s. | <0.001 | n.s. |
| Fasting glucose (mmol/L) | 5.64 ± 0.12 | 5.46 ± 0.14 | 6.86 ± 0.09 | 6.32 ± 0.16 ** | <0.001 | <0.05 | n.s. |
| Non-fasting glucose (mmol/L) | 6.74 ± 0.04 | 6.86 ± 0.13 | 8.68 ± 0.28 | 8.01 ± 0.18 * | ˂0.001 | n.s. | <0.05 |
| Insulin (nmol/L) | 0.252 ± 0.034 | 0.176 ± 0.016 * | 0.299 ± 0.019 | 0.125 ± 0.014 *** | n.s. | <0.001 | <0.05 |
| Glucagon (pg/mL) | 300.7 ± 40.4 | 267.6 ± 19.7 | 227.7 ± 5.2 | 240.3 ± 18.9 | n.s. | n.s. | n.s. |
| AUC0-180 (mmol/L) | 1303 ± 21 | 1227 ± 11 * | 1604 ± 22 | 1507 ± 26 ** | <0.001 | <0.001 | n.s. |
| Serum TAG (mmol/L) | 1.20 ± 0.16 | 0.84 ± 0.12 | 4.86 ± 0.25 | 3.55 ± 0.26 *** | <0.001 | <0.001 | <0.05 |
| Serum cholesterol (mmol/L) | 2.03 ± 0.11 | 2.08 ± 0.13 | 2.05 ± 0.05 | 1.90 ± 0.06 | n.s. | n.s. | n.s. |
| HDL-cholesterol (mmol/L) | 0.97 ± 0.06 | 1.00 ± 0.09 | 0.94 ± 0.03 | 0.92 ± 0.02 | n.s. | n.s. | n.s. |
| NEFA (mmol/L) | 0.63 ± 0.04 | 0.64 ± 0.02 | 0.80 ± 0.05 | 0.79 ± 0.06 | <0.01 | n.s. | n.s. |
| HMW adiponectin (ng/mL) | 1.10 ± 0.07 | 1.09 ± 0.08 | 1.00 ± 0.09 | 1.05 ± 0.04 | n.s. | n.s. | n.s. |
| Leptin (ng/mL) | 9.83 ± 0.93 | 6.23 ± 0.80 ** | 14.12 ± 0.91 | 7.39 ± 0.64 *** | <0.01 | <0.001 | n.s. |
| MCP-1 (pmol/L) | 4.89 ± 0.20 | 4.30 ± 0.16 * | 6.20 ± 0.18 | 5.50 ± 0.20 * | <0.001 | <0.01 | n.s. |
| TNFα (pg/mL) | 2.33 ± 0.24 | 2.36 ± 0.26 | 3.13 ± 0.23 | 3.33 ± 0.31 | <0.01 | n.s. | n.s. |
| IL-6 (pg/mL) | 91.96 ± 4.82 | 83.32 ± 4.88 | 114.04 ± 5.87 | 126.74 ± 6.93 | <0.001 | n.s. | n.s. |
| hsCRP (μg/mL) | 1.94 ± 0.22 | 1.60 ± 0.18 | 1.49 ± 0.09 | 1.74 ± 0.16 | n.s. | n.s. | n.s. |
| β-hydroxybutyrate (μmol/L) | 1.00 ± 0.12 | 0.83 ± 0.09 | 1.13 ± 0.09 | 1.11 ± 0.09 | <0.05 | n.s. | n.s. |
| Wistar | Wistar + Empa | HHTg | HHTg + Empa | PS | PT | PI | |
|---|---|---|---|---|---|---|---|
| Relative liver weight (mg/g) | 2.42 ± 0.06 | 2.37 ± 0.05 | 3.15 ± 0.08 | 3.11 ± 0.06 | ˂0.001 | n.s. | n.s. |
| TAG in the liver (μmol/g) | 8.21 ± 0.39 | 7.71 ± 0.36 | 11.79 ± 0.44 | 9.67 ± 0.29 *** | <0.001 | <0.01 | <0.05 |
| DAG in the liver (μmol/g) | 1.85 ± 0.07 | 1.30 ± 0.09 *** | 2.27 ± 0.12 | 1.39 ± 0.07 *** | <0.01 | ˂0.001 | n.s. |
| Cholesterol in the liver (μmol/g) | 12.04 ± 0.59 | 12.29 ± 0.56 | 13.68 ± 0.36 | 14.20 ± 0.35 | <0.001 | n.s. | n.s. |
| Glycogen in the liver (μmol/g) | 188.75 ± 20.08 | 84.12 ± 8.67 *** | 315.68 ± 24.03 | 223.14 ± 14.89 *** | <0.001 | <0.001 | n.s. |
| GSH/GSSG (μmol/g) | 23.17 ± 1.81 | 24.57 ± 3.25 | 16.39 ± 0.87 | 22.89 ± 1.25 * | <0.05 | n.s. | n.s. |
| SOD (U/mg) | 0.103 ± 0.006 | 0.103 ± 0.016 | 0.093 ± 0.009 | 0.173 ± 0.016 *** | <0.05 | <0.001 | <0.001 |
| GPx (μM NADPH/min/mg) | 224.9 ± 22.7 | 275.6 ± 33.1 | 149.3 ± 18.5 | 234.5 ± 16.3 * | <0.05 | <0.01 | n.s. |
| MDA (nmol/mg) | 3.82 ± 0.38 | 3.47 ± 0.46 | 3.92 ± 0.37 | 3.38 ± 0.72 | n.s. | n.s. | n.s. |
| 4-HNE (nmol/mg) | 0.61 ± 0.05 | 0.63 ± 0.04 | 0.53 ± 0.10 | 0.52 ± 0.03 | n.s. | n.s. | n.s. |
| Serum ALT (μkat/L) | 1.05 ± 0.09 | 1.25 ± 0.17 | 1.28 ± 0.10 | 1.36 ± 0.13 | n.s. | n.s. | n.s. |
| Serum AST (μkat/L) | 2.62 ± 0.11 | 2.76 ± 0.15 | 3.34 ± 0.21 | 3.25 ± 0.14 | ˂0.01 | n.s. | n.s. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hüttl, M.; Markova, I.; Miklankova, D.; Zapletalova, I.; Poruba, M.; Haluzik, M.; Vaněčkova, I.; Malinska, H. In a Prediabetic Model, Empagliflozin Improves Hepatic Lipid Metabolism Independently of Obesity and before Onset of Hyperglycemia. Int. J. Mol. Sci. 2021, 22, 11513. https://doi.org/10.3390/ijms222111513
Hüttl M, Markova I, Miklankova D, Zapletalova I, Poruba M, Haluzik M, Vaněčkova I, Malinska H. In a Prediabetic Model, Empagliflozin Improves Hepatic Lipid Metabolism Independently of Obesity and before Onset of Hyperglycemia. International Journal of Molecular Sciences. 2021; 22(21):11513. https://doi.org/10.3390/ijms222111513
Chicago/Turabian StyleHüttl, Martina, Irena Markova, Denisa Miklankova, Iveta Zapletalova, Martin Poruba, Martin Haluzik, Ivana Vaněčkova, and Hana Malinska. 2021. "In a Prediabetic Model, Empagliflozin Improves Hepatic Lipid Metabolism Independently of Obesity and before Onset of Hyperglycemia" International Journal of Molecular Sciences 22, no. 21: 11513. https://doi.org/10.3390/ijms222111513
APA StyleHüttl, M., Markova, I., Miklankova, D., Zapletalova, I., Poruba, M., Haluzik, M., Vaněčkova, I., & Malinska, H. (2021). In a Prediabetic Model, Empagliflozin Improves Hepatic Lipid Metabolism Independently of Obesity and before Onset of Hyperglycemia. International Journal of Molecular Sciences, 22(21), 11513. https://doi.org/10.3390/ijms222111513