
Neutrophil Extracellular Traps (NETs) and Hypercoagulability in Plasma Cell Dyscrasias—Is This Phenomenon Worthy of Exploration?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- –
- Accumulation of light immunoglobulin chains in distal tubules, forming obstructive casts;
- –
- Direct toxicity of light chains to proximal tubules, where they are endocytosed and induce necrosis of tubule cells; and
- –
- Hypercalcemia, decreasing renal concentrating ability [5].
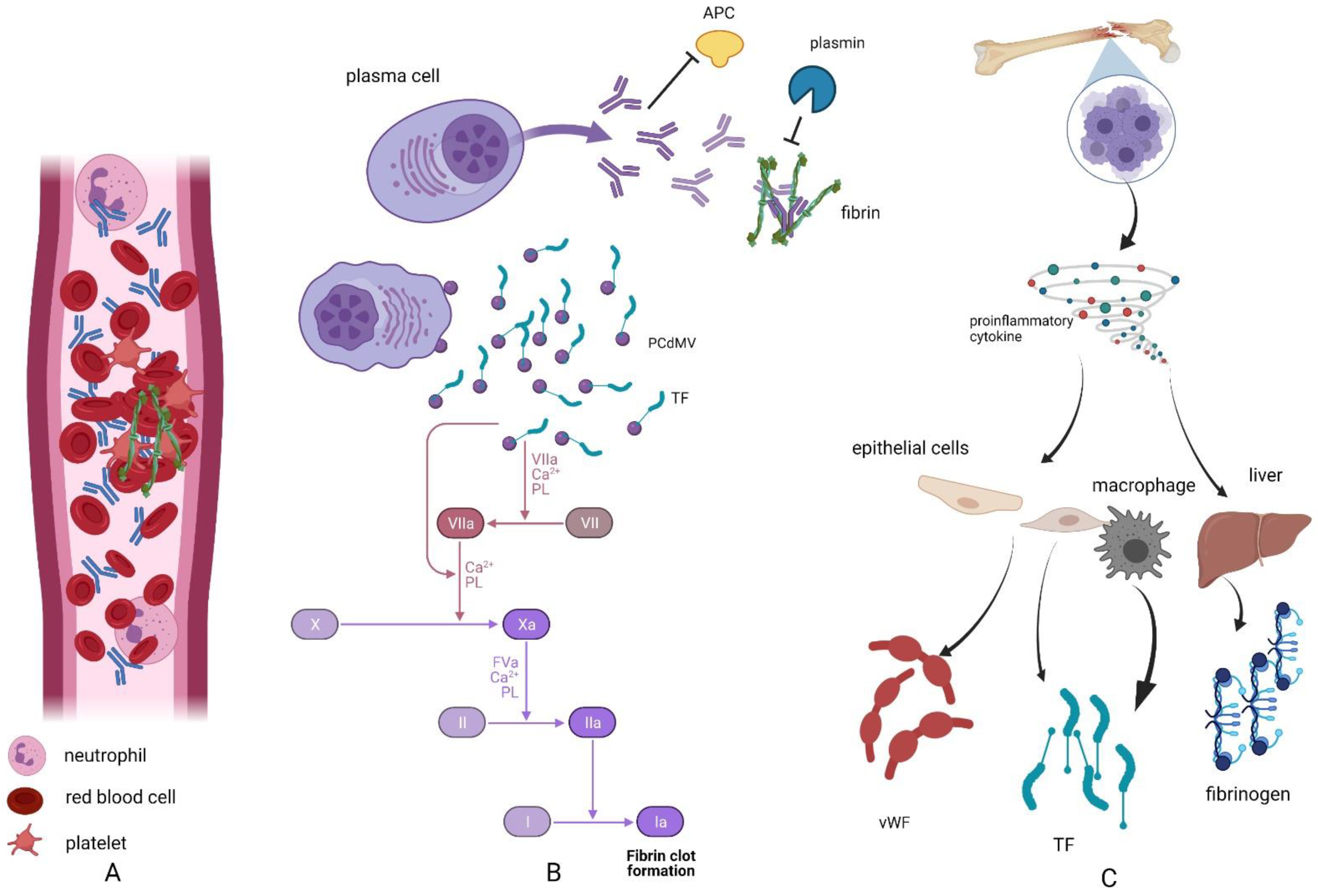
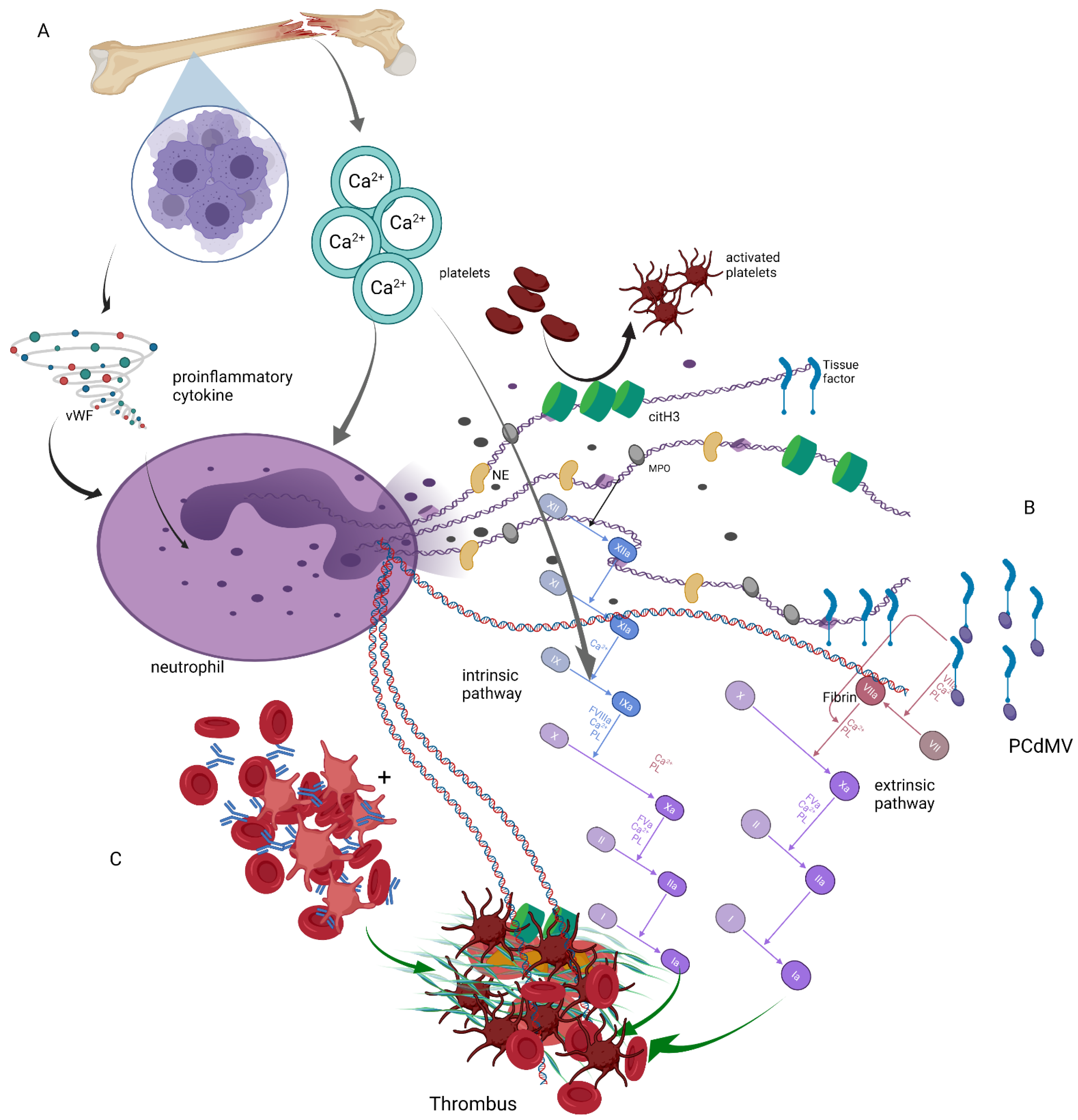
2. Hypercoagulability in Plasma Cell Dyscrasias
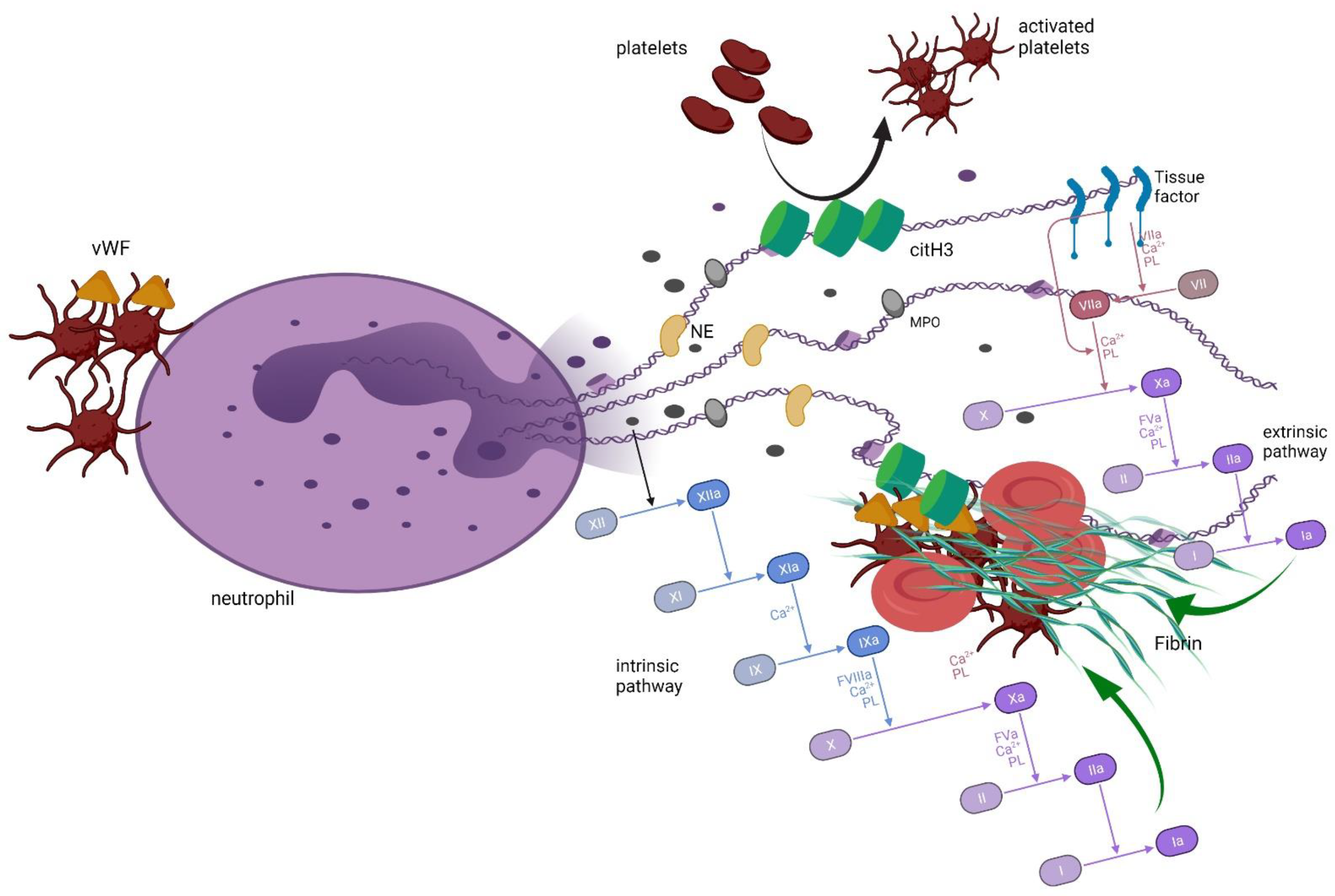
3. Neutrophil Extracellular Traps and Coagulation
4. Neutrophil Extracellular Traps in Plasma Cell Dyscrasias
5. Summary
Funding
Conflicts of Interest
References
- Soh, K.T.; Tario, J.D.; Wallace, P.K. Diagnosis of Plasma Cell Dyscrasias and Monitoring of Minimal Residual Disease by Multiparametric Flow Cytometry. Clin. Lab. Med. 2017, 37, 821–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannopoulos, K.; Usnarska-Zubkiewicz, L.; Dytfeld, D.; Jurczyszyn, A.; Walewski, J.; Lech-Marańda, E.; Walter-Croneck, A.; Pieńkowska-Grela, B.; Wróbel, T.; Charliński, G.; et al. Zalecenia Polskiej Grupy Szpiczakowej Dotyczące Rozpoznawania i Leczenia Szpiczaka Plazmocytowego Oraz Innych Dyskrazji Plazmocytowych na rok 2021; Polska Grupa Szpiczakowa: Lublin, Poland, 2021. [Google Scholar]
- Dimopoulos, M.A.; Moreau, P.; Terpos, E.; Mateos, M.-V.; Zweegman, S.; Cook, G.; Delforge, M.; Hájek, R.; Schjesvold, F.; Cavo, M.; et al. Multiple Myeloma: EHA-ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up. Ann. Oncol. 2021, 32, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Kwaan, H.C. Hyperviscosity in plasma cell dyscrasias. Clin. Hemorheol. Microcirc. 2013, 55, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.; Kastritis, E.; Rosiñol, L.; Blade, J.; Ludwig, H.; Blad, J. Pathogenesis and treatment of renal failure in multiple myeloma. Leukemia 2008, 22, 1485–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclerc, V.; Karlin, L.; Herledan, C.; Marchal, L.; Baudouin, A.; Gouraud, A.; Caffin, A.G.; Larbre, V.; Lazareth, A.; Bachy, E.; et al. Thromboembolic events and thromboprophylaxis associated with immunomodulators in multiple myeloma patients: A real-life study. J. Cancer Res. Clin. Oncol. 2021, 1–10. [Google Scholar] [CrossRef]
- Papageorgiou, L.; Hussen, K.A.; Thouroude, S.; Mbemba, E.; Cost, H.; Garderet, L.; Elalamy, I.; Larsen, A.; Van Dreden, P.; Dimopoulos, M.A.; et al. Modelization of Blood-Borne Hypercoagulability in Myeloma: A Tissue-Factor-Bearing Microparticle-Driven Process. TH Open 2019, 3, e340–e347. [Google Scholar] [CrossRef] [Green Version]
- Cornell, R.F.; Goldhaber, S.Z.; Engelhardt, B.G.; Moslehi, J.; Jagasia, M.; Harrell, S.; Rubinstein, S.M.; Hall, R.; Wyatt, H.; Piazza, G. Primary prevention of venous thromboembolism with apixaban for multiple myeloma patients receiving immunomodulatory agents. Br. J. Haematol. 2020, 190, 555–561. [Google Scholar] [CrossRef]
- Baccouche, H.; Hadhri, M.; Aissi, W.; Chakroun, A.; Bahri, D.; Mahjoub, S.; Ben Romdhane, N. The hypercoagulable state in multiple myeloma: The contribution of thrombin generation test. Int. J. Lab. Hematol. 2019, 41, 684–690. [Google Scholar] [CrossRef]
- Fotiou, D.; Gavriatopoulou, M.; Terpos, E. Multiple Myeloma and Thrombosis: Prophylaxis and Risk Prediction Tools. Cancers 2020, 12, 191. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, T.; Kristensen, S.R.; Gregersen, H.; Teodorescu, E.M.; Pedersen, S. Prothrombotic abnormalities in patients with multiple myeloma and monoclonal gammopathy of undetermined significance. Thromb. Res. 2021, 202, 108–118. [Google Scholar] [CrossRef]
- De Stefano, V.; Za, T.; Rossi, E. Venous thromboembolism in multiple myeloma. Semin. Thromb. Hemost. 2014, 40, 338–347. [Google Scholar] [CrossRef]
- Auwerda, J.J.; Sonneveld, P.; de Maat, M.P.; Leebeek, F.W. Prothrombotic Coagulation Abnormalities in Patients with Paraprotein-Producing B-Cell Disorders. Clin. Lymphoma Myeloma 2007, 7, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Reddel, C.J.; Tan, C.W.; Chen, V.M. Thrombin Generation and Cancer: Contributors and Consequences. Cancers 2019, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppenbrouwers, T.; Autar, A.S.A.; Sultan, A.R.; Abraham, T.E.; Van Cappellen, W.A.; Houtsmuller, A.B.; Van Wamel, W.J.B.; Van Beusekom, H.M.M.; Van Neck, J.W.; De Maat, M.P.M. In vitro induction of NETosis: Comprehensive live imaging comparison and systematic review. PLoS ONE 2017, 12, e0176472. [Google Scholar] [CrossRef] [Green Version]
- Bawadekar, M.; Shim, D.; Johnson, C.J.; Warner, T.F.; Rebernick, R.; Damgaard, D.; Nielsen, C.H.; Pruijn, G.J.; Nett, J.E.; Shelef, M.A. Peptidylarginine deiminase 2 is required for tumor necrosis factor alpha-induced citrullination and arthritis, but not neutrophil extracellular trap formation. J. Autoimmun. 2017, 80, 39–47. [Google Scholar] [CrossRef]
- Manda-Handzlik, A.; Bystrzycka, W.; Wachowska, M.; Sieczkowska, S.; Stelmaszczyk-Emmel, A.; Demkow, U.; Ciepiela, O. The influence of agents differentiating HL-60 cells toward granulocyte-like cells on their ability to release neutrophil extracellular traps. Immunol. Cell Biol. 2018, 96, 413–425. [Google Scholar] [CrossRef] [PubMed]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef]
- Thalin, C.; Hisada, Y.; Lundstrom, S.; Mackman, N.; Wallen, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Thålin, C.; Demers, M.; Blomgren, B.; Wong, S.L.; von Arbin, M.; von Heijne, A.; Laska, A.C.; Wallén, H.; Wagner, D.D.; Aspberg, S. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb. Res. 2016, 139, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Sun, W.; Cui, W.; Li, X.; Yao, J.; Jia, X.; Li, C.; Wu, H.; Hu, Z.; Zou, X. Procoagulant role of neutrophil extracellular traps in patients with gastric cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 14075–14086. [Google Scholar]
- Hisada, Y.; Geddings, J.E.; Ay, C.; Mackman, N. Venous thrombosis and cancer: From mouse models to clinical trials. J. Thromb. Haemost. 2015, 13, 1372–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Zhang, Y.; Cao, M.; Li, T.; Yao, Z.; Zhao, L.; Fang, S.; Yu, B.; Dong, Z.; Thatte, H.S.; et al. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thromb. Haemost. 2016, 115, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef] [PubMed]
- Chornenki, N.L.J.; Dwivedi, D.J.; Kwong, A.C.; Zamir, N.; Fox-Robichaud, A.E.; Liaw, P.C. The Canadian Critical Care Translational Biology Group Identification of hemostatic markers that define the pre-DIC state: A multi-center observational study. J. Thromb. Haemost. 2020, 18, 2524–2531. [Google Scholar] [CrossRef] [PubMed]
- Kenny, M.; Schoen, I. A handshake between platelets and neutrophils might fuel deep vein thrombosis. Platelets 2020, 31, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Yang, L.; Braun, A.; Anders, H.-J. Extracellular DNA—A Danger Signal Triggering Immunothrombosis. Front. Immunol. 2020, 11, 568513. [Google Scholar] [CrossRef]
- McDonald, B.; Davis, R.P.; Kim, S.-J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, S.; Opneja, A.; Nayak, L. The role of neutrophils in thrombosis. Thromb. Res. 2018, 170, 87–96. [Google Scholar] [CrossRef]
- Zucoloto, A.Z.; Jenne, C.N. Platelet-Neutrophil Interplay: Insights into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 2019, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Elaskalani, O.; Razak, N.B.A.; Metharom, P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun. Signal. 2018, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.-B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Varjú, I.; Kolev, K. Networks that stop the flow: A fresh look at fibrin and neutrophil extracellular traps. Thromb. Res. 2019, 182, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stavrou, E.X.; Fang, C.; Bane, K.L.; Long, A.T.; Naudin, C.; Kucukal, E.; Gandhi, A.; Brett-Morris, A.; Mumaw, M.M.; Izadmehr, S.; et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J. Clin. Investig. 2018, 128, 944–959. [Google Scholar] [CrossRef] [Green Version]
- Ducroux, C.; Di Meglio, L.; Loyau, S.; Delbosc, S.; Boisseau, W.; Deschildre, C.; Ben Maacha, M.; Blanc, R.; Redjem, H.; Ciccio, G.; et al. Thrombus Neutrophil Extracellular Traps Content Impair tPA-Induced Thrombolysis in Acute Ischemic Stroke. Stroke 2018, 49, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lin, C.; Leso, A.; Nefedova, Y. Quantification of Citrullinated Histone H3 Bound DNA for Detection of Neutrophil Extracellular Traps. Cancers 2020, 12, 3424. [Google Scholar] [CrossRef]
- Fagerhol, M.K.; Johnson, E.; Tangen, J.; Hollan, I.; Mirlashari, M.R.; Nissen-Meyer, L.S.H.; Hetland, G. NETs analysed by novel calprotectin-based assays in blood donors and patients with multiple myeloma or rheumatoid arthritis: A pilot study. Scand. J. Immunol. 2020, 91, e12870. [Google Scholar] [CrossRef]
- Meijer, B.; Gearry, R.B.; Day, A.S. The Role of S100A12 as a Systemic Marker of Inflammation. Int. J. Inflamm. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Lin, C.; Deng, H.; Strnad, J.; Bernabei, L.; Vogl, D.T.; Burke, J.J.; Nefedova, Y. A Novel Peptidylarginine Deiminase 4 (PAD4) Inhibitor BMS-P5 Blocks Formation of Neutrophil Extracellular Traps and Delays Progression of Multiple Myeloma. Mol. Cancer Ther. 2020, 19, 1530–1538. [Google Scholar] [CrossRef]
- Lauw, M.N.; Leebeek, F.W.; De Maat, M.P.; Van Mierlo, G.J.; Middeldorp, S.; Zeerleder, S.S. Elevated Levels of Circulating Nucleosomes Are Not Associated with Venous Thrombosis or Neutrophil Activation in Patients with Multiple Myeloma. Blood 2016, 128, 274. [Google Scholar] [CrossRef]
- Azevedo, E.P.C.; Guimarães-Costa, A.B.; Torezani, G.S.; Braga, C.A.; Palhano, F.L.; Kelly, J.W.; Saraiva, E.M.; Foguel, D. Amyloid Fibrils Trigger the Release of Neutrophil Extracellular Traps (NETs), Causing Fibril Fragmentation by NET-associated Elastase. J. Biol. Chem. 2012, 287, 37206–37218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Aymonnier, K.; Wagner, D.D. Neutrophil stimulation with citrullinated histone H4 slows down calcium influx and reduces NET formation compared with native histone H4. PLoS ONE 2021, 16, e0251726. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry (Moscow) 2020, 85, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Maueröder, C.; Hirth, S.; Nowecki, S.; Günther, C.; Billmeier, U.; Paulus, S.; Biermann, M.; Munoz, L.E.; Hoffmann, M.; et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat. Commun. 2016, 7, 10973. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciepiela, O.; Małecka-Giełdowska, M.; Czyżewska, E. Neutrophil Extracellular Traps (NETs) and Hypercoagulability in Plasma Cell Dyscrasias—Is This Phenomenon Worthy of Exploration? J. Clin. Med. 2021, 10, 5243. https://doi.org/10.3390/jcm10225243
Ciepiela O, Małecka-Giełdowska M, Czyżewska E. Neutrophil Extracellular Traps (NETs) and Hypercoagulability in Plasma Cell Dyscrasias—Is This Phenomenon Worthy of Exploration? Journal of Clinical Medicine. 2021; 10(22):5243. https://doi.org/10.3390/jcm10225243
Chicago/Turabian StyleCiepiela, Olga, Milena Małecka-Giełdowska, and Emilia Czyżewska. 2021. "Neutrophil Extracellular Traps (NETs) and Hypercoagulability in Plasma Cell Dyscrasias—Is This Phenomenon Worthy of Exploration?" Journal of Clinical Medicine 10, no. 22: 5243. https://doi.org/10.3390/jcm10225243