PD-L1 Expression in Patients with Idiopathic Pulmonary Fibrosis


Abstract
:1. Introduction
2. Methods
2.1. Patients
2.2. Data Collection
2.3. Cryobiopsy Procedure
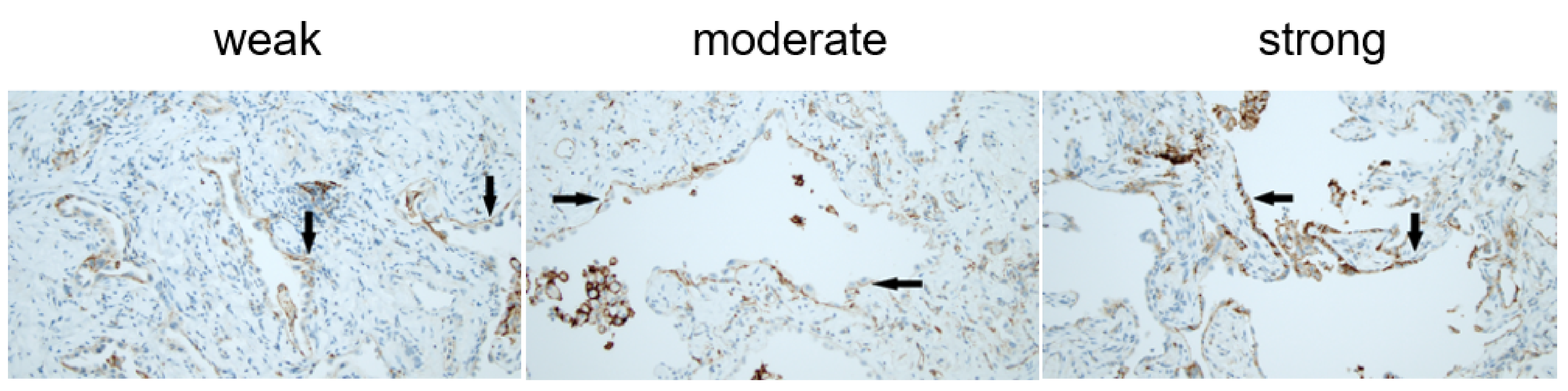
2.4. Pathology Method
2.5. Statistics
3. Results
3.1. Demographics
3.2. Specific Histological Lesions
3.3. One-Year Follow-Up
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, S.-A.W.; Dobelle, M.; Padilla, M.; Agovino, M.; Wisnivesky, J.P.; Hashim, D.; Boffetta, P. Idiopathic Pulmonary Fibrosis and Lung Cancer. A Systematic Review and Meta-analysis. Ann. Am. Thorac. Soc. 2019, 16, 1041–1051. [Google Scholar] [CrossRef]
- Jafarinezhad, A.; YektaKooshali, M.H. Lung cancer in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0202360. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Gomatou, G.; Bouros, E.; Trigidou, R.; Tzilas, V.; Bouros, D. Common Pathogenic Mechanisms Between Idiopathic Pulmonary Fibrosis and Lung Cancer. Chest 2019, 156, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Caliò, A.; Lever, V.; Rossi, A.; Gilioli, E.; Brunelli, M.; Dubini, A.; Tomassetti, S.; Piciucchi, S.; Nottegar, A.; Rossi, G.; et al. Increased frequency of bronchiolar histotypes in lung carcinomas associated with idiopathic pulmonary fibrosis. Histopathology 2017, 71, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Nakajima, T.; Matsusaka, K.; Fukuyo, M.; Nakayama, M.; Morimoto, J.; Ito, Y.; Yamamoto, T.; Sakairi, Y.; Rahmutulla, B.; et al. Genetic alterations in squamous cell lung cancer associated with idiopathic pulmonary fibrosis. Int. J. Cancer 2021, 148, 3008–3018. [Google Scholar] [CrossRef] [PubMed]
- Taskar, V.; Coultas, D. Exposures and idiopathic lung disease. Semin. Respir. Crit. Care Med. 2008, 29, 670–679. [Google Scholar] [CrossRef]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 2006, 3, 293–298. [Google Scholar] [CrossRef]
- Spira, A.; Beane-Ebel, J.; Shah, V.; Liu, G.; Schembri, F.; Yang, X.; Palma, J.; Brody, J.S. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc. Natl. Acad. Sci. USA 2004, 101, 10143–10148. [Google Scholar] [CrossRef] [Green Version]
- Carloni, A.; Poletti, V.; Fermo, L.; Bellomo, N.; Chilosi, M. Heterogeneous distribution of mechanical stress in human lung: A mathematical approach to evaluate abnormal remodeling in IPF. J. Theor. Biol. 2013, 332, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Fingerlin, T.E.; Evans, C.M.; Schwarz, M.I.; Schwartz, D.A. MUC5B and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 2), S193–S199. [Google Scholar]
- Moore, C.; Blumhagen, R.Z.; Yang, I.V.; Walts, A.; Powers, J.; Walker, T.; Bishop, M.; Russell, P.; Vestal, B.; Cardwell, J.; et al. Resequencing Study Confirms That Host Defense and Cell Senescence Gene Variants Contribute to the Risk of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 199–208. [Google Scholar] [CrossRef]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef]
- Renzoni, E.; Poletti, V.; Mackintosh, J.A. Disease pathology in fibrotic interstitial lung disease:is it all about usual interstitial pneumonia? Lancet 2021, 398, 1437–1449. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Zamò, A.; Doglioni, C.; Reghellin, D.; Lestani, M.; Montagna, L.; Pedron, S.; Ennas, M.G.; Cancellieri, A.; Murer, B.; et al. Migratory marker expression in fibroblast foci of idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Nie, R.-C.; Zhao, C.-B.; Xia, X.-W.; Luo, Y.-S.; Wu, T.; Zhou, Z.-W.; Yuan, S.-Q.; Wang, Y.; Li, Y.-F. The Efficacy and Safety of PD-1/PD-L1 Inhibitors in Combination with Conventional Therapies for Advanced Solid Tumors: A Meta-Analysis. BioMed Res. Int. 2020, 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sznol, M.; Chen, L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin. Cancer Res. 2013, 19, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Larsen, T.V.; Hussmann, D.; Nielsen, A.L. PD-L1 and PD-L2 expression correlated genes in non-small-cell lung cancer. Cancer Commun. 2019, 39, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2021, 7, e1423170. [Google Scholar] [CrossRef] [Green Version]
- Salton, F.; Volpe, M.; Confalonieri, M. Epithelial–Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovic, D.; Milenkovic, M.R.; Stevuljevic, J.K.; Markovic, J.; Ceriman, V.; Kontic, M.; Trifunovic, V.S. Membrane PD-L1 expression and soluble PD-L1 plasma levels in idiopathic pulmonary fibrosis—a pilot study. J. Thorac. Dis. 2018, 10, 6660–6669. [Google Scholar] [CrossRef] [Green Version]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, K.; Liu, M.; Zheng, J.; Wen, L.; Chen, Q.; Xiang, Z.; Lam, K.-T.; Liu, Y.; Chan, G.C.-F.; Lau, Y.-L.; et al. PD-1/PD-L1 Pathway Mediates the Alleviation of Pulmonary Fibrosis by Human Mesenchymal Stem Cells in Humanized Mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Stanojevic, S.; Graham, B.L.; Cooper, B.; Thompson, B.R.; Carter, K.W.; Francis, R.W.; Hall, G. Official ERS technical standards: Global Lung Function Initiative reference values for the carbon monoxide transfer factor for Caucasians. Eur. Respir. J. 2017, 50, 1700010. [Google Scholar] [CrossRef]
- Kronborg-White, S.; Sritharan, S.S.; Madsen, L.B.; Folkersen, B.; Voldby, N.; Poletti, V.; Rasmussen, T.R.; Bendstrup, E. Integration of cryobiopsies for interstitial lung disease diagnosis is a valid and safe diagnostic strategy—Experiences based on 250 biopsy procedures. J. Thorac. Dis. 2021, 13, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Ravaglia, C.; Rossi, G.; Tomassetti, S.; Dubini, A.; Piciucchi, S.; Chilosi, M. Report Standardization in Transbronchial Lung Cryobiopsy. Arch. Pathol. Lab. Med. 2019, 143, 416–417. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M.; Hirai, S.; Tanaka, Y.; Sumi, T.; Miyajima, M.; Mishina, T.; Yamada, G.; Otsuka, M.; Hasegawa, T.; Kojima, T.; et al. Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial–mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab. Investig. 2016, 97, 232–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [Green Version]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis—Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Neuser, P.; Prasse, A.; Kreuter, M.; Rabe, K.; Schade-Brittinger, C.; Wagner, J.; Günther, A. Exploring efficacy and safety of oral Pirfenidone for progressive, non-IPF lung fibrosis (RELIEF)—A randomized, double-blind, placebo-controlled, parallel group, multi-center, phase II trial. BMC Pulm. Med. 2017, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Maher, T.M.; Corte, T.J.; Fischer, A.; Kreuter, M.; Lederer, D.J.; Molina-Molina, M.; Axmann, J.; Kirchgaessler, K.-U.; Samara, K.; Gilberg, F.; et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2020, 8, 147–157. [Google Scholar] [CrossRef]
- Khunger, M.; Rakshit, S.; Pasupuleti, V.; Hernandez, A.V.; Mazzone, P.; Stevenson, J. Incidence of Pneumonitis with Use of Programmed Death 1 and Programmed Death-Ligand 1 Inhibitors in Non-Small Cell Lung Cancer: A systematic review and meta-analysis of trials. Chest 2017, 152, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atchley, W.T.; Alvarez, C.; Saxena-Beem, S.; Schwartz, T.A.; Ishizawar, R.C.; Patel, K.P. Immune Checkpoint Inhibitor-Related Pneumonitis in Lung Cancer: Real-World Incidence, Risk Factors, and Management Practices Across Six Health-Care Centers in North Carolina. Chest 2021, 160, 731–742. [Google Scholar] [CrossRef]
- Duchemann, B.; Didier, M.; Pailler, M.-C.; Brillet, P.-Y.; Kambouchner, M.; Uzunhan, Y. Can nivolumab be used safely in idiopathic pulmonary fibrosis? Rev. Mal. Respir. 2019, 36, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Ide, M.; Tanaka, K.; Sunami, S.; Asoh, T.; Maeyama, T.; Tsuruta, N.; Nakanishi, Y.; Okamoto, I. Durable response to nivolumab in a lung adenocarcinoma patient with idiopathic pulmonary fibrosis. Thorac. Cancer 2018, 9, 1519–1521. [Google Scholar] [CrossRef]
- Fujimoto, D.; Yomota, M.; Sekine, A.; Morita, M.; Morimoto, T.; Hosomi, Y.; Ogura, T.; Tomioka, H.; Tomii, K. Nivolumab for advanced non-small cell lung cancer patients with mild idiopathic interstitial pneumonia: A multicenter, open-label single-arm phase II trial. Lung Cancer 2019, 134, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, M.; Prévot, G.; Collot, S.; Guilleminault, L.; Didier, A.; Mazières, J. Management of pulmonary toxicity associated with immune checkpoint inhibitors. Eur. Respir. Rev. 2019, 28, 190012. [Google Scholar] [CrossRef] [PubMed]
- Hakroush, S.; Kopp, S.B.; Tampe, D.; Gersmann, A.-K.; Korsten, P.; Zeisberg, M.; Tampe, B. Variable Expression of Programmed Cell Death Protein 1-Ligand 1 in Kidneys Independent of Immune Checkpoint Inhibition. Front. Immunol. 2021, 11, 3546. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| IPF Patients 43 (44%) | Non-IPF Patients 55 (56%) | |
|---|---|---|
| Age (median, range) | 73 (48–79) * | 70 (40–78) * |
| Gender (male) | 26 (60%) | 34 (62%) |
| Smoking % | ||
| 35 | 36 |
| 56 | 51 |
| 9 | 13 |
| Packyears (SD) | 30.21 ± 19 | 25.24 ± 18 |
| FVC % of predicted (SD) | 93.60 ± 19 | 92.73 ± 21 |
| TLco % of predicted (median, IQR) | 60 (49–70) | 55 (49–66) |
| 6MWT distance (m) (SD) | 466 ± 96 | 491 ± 99 |
| Desaturation ≥ 4 % during 6MWT (SD) | 35 (83%) ** | 35 (71%) ** |
| Traction bronchiectasis | 41 (95%) * | 35 (64%) * |
| GAP index | ||
| 34 (79%) | 46 (84%) |
| 9 (21%) | 9 (16%) |
| IPF mPD-L1pos n = 12 | Non-IPF mPD-L1pos n = 5 | |
|---|---|---|
| Age (median, range) | 76 (67–77) | 72 (64–75) |
| Gender (male) | 6 (50%) | 3 (60%) |
| Smoking % | ||
| 58 | 40 |
| 33 | 40 |
| 8 | 20 |
| Packyears (SD) | 31 ± 15 | 37.67 ± 27 |
| FVC % of predicted (SD) | 92.31 ± 19 | 99.83 ± 11 |
| TLco% of predicted (median, IQR) | 51 (46–61) | 53 (50–61) |
| 6MWT distance (m) (SD) | 470 ± 86 | 506 ± 19 |
| Desaturation ≥ 4 % during 6MWT | 9 (75%) | 3 (60%) |
| Traction bronchiectasis | 12 (100%) * | 2 (40%) * |
| GAP index | ||
| 7 (58%) | 5 (100%) |
| 5 (42%) | 0 |
| mPD-L1pos n = 17 | mPD-L1neg n = 81 | |
|---|---|---|
| Age (median, range) | 68 (48–79) | 68 (40–79) |
| Gender (male) | 9 (53%) | 51 (63%) |
| Smoking % | ||
| 53 | 32 |
| 35 | 57 |
| 12 | 11 |
| Packyears (SD) | 33.5 ± 18 | 26.53 ± 21 |
| FVC, % of predicted (SD) | 93.29 ± 17 | 90.52 ± 20 |
| TLco, % of predicted (median, IQR) | 51 (47–61) | 60 (49–68) |
| 6MWT distance (m) (SD) | 481 ± 74 | 479 ± 103 |
| Desaturation ≥ 4 % during 6MWT | 12 (71%) ** | 58 (82%) ** |
| Traction bronchiectasis | 14 (82%) | 62 (77%) |
| GAP index | ||
| 12 (71%) | 68 (84%) |
| 5 (29%) | 13 (16%) |
| IPF mPD-L1pos n = 12 | IPF mPD-L1neg n = 31 | |
|---|---|---|
| Age (median, range) | 76 (67–77) | 72 (64–75) |
| Gender (male) | 6 (50%) | 20 (64%) |
| Smoking % | ||
| 58 | 26 |
| 33 | 64 |
| 8 | 10 |
| Packyears | 31 ± 15 | 30.03 ± 21 |
| FVC, % of predicted (SD) | 91.33 ± 18 | 94.03 ± 22 |
| TLco, % of predicted (median, IQR) | 51 (46–61) * | 63 (55–72) * |
| 6MWT distance (m) (SD) | 470 ± 86 | 464 ± 101 |
| Desaturation ≥ 4 % during 6MWT | 9 (75%) ** | 26 (87%) ** |
| Traction bronchiectasis | 12 (100%) | 29 (94%) |
| GAP index | ||
| 7 (58%) * | 27 (87%) * |
| 5 (42%) * | 4 (13%) * |
| IPF Patients n = 43 | Non- IPF Patients n = 55 | p-Value | |
| mPD-L1 | 12 (28%) | 5 (9%) | 0.015 |
| gPD-L1 | 33 (76%) | 48 (89%) | 0.172 |
| Fibroblast foci | 17 (40%) | 13 (24%) | 0.09 |
| Honeycombing | 9 (21%) | 2 (4%) | 0.007 |
| Patchy fibrosis | 41 (95%) | 43 (78%) | 0.016 |
| Patchy fibrosis + fibroblast foci | 17 (40%) | 12 (22%) | 0.057 |
| mPD-L1pos n = 17 | mPD-L1neg n = 81 | p-Value | |
| Fibroblast foci | 9 (53%) | 21 (26%) | 0.028 |
| Honeycombing | 2 (12%) | 9 (11%) | 0.938 |
| Patchy fibrosis | 17 (100%) | 67 (83%) | 0.064 |
| Patchy fibrosis + fibroblast foci | 9 (53%) | 20 (25%) | 0.02 |
| IPF mPD-L1pos n = 12 | Non-IPF mPD-L1pos n = 5 | p-Value | |
| Fibroblast foci | 5 (42%) | 4 (80%) | 0.149 |
| Honeycombing | 2 (17%) | 0 | 0.331 |
| Patchy fibrosis | 12 (100%) | 5 (100%) | |
| Patchy fibrosis + fibroblast foci | 5 (42%) | 4 (80%) | 0.149 |
| IPF mPD-L1pos n = 12 | IPF mPD-L1neg n = 31 | p-Value | |
| Fibroblast foci | 5 (42%) | 12 (39%) | 0.859 |
| Honeycombing | 2 (17%) | 7 (23%) | 0.669 |
| Patchy fibrosis | 12 (100%) | 29 (94%) | 0.368 |
| Patchy fibrosis + fibroblast foci | 5 (42%) | 12 (39%) | 0.859 |
| IPF n = 41 | Non-IPF n = 50 | p-Value | |
| Change FVC % (SD) | −3.3 ± 10 | −1.64 ± 13 | 0.49 |
| Change TLco % (SD) | −1.5 ± 8 | 5.2 ± 12 | 0.003 |
| Change 6MWTD (m) median (IQR) | −12 (−42–13) | −25 (−59–36) | 0.14 |
| mPD-L1pos n = 15 | mPD-L1neg n = 76 | p-Value | |
| Change FVC % (SD) | −1.3 ± 11 | −2.6 ± 12 | 0.68 |
| Change TLco % (SD) | 0.6 ± 9 | 2.5 ± 11 | 0.53 |
| Change 6MWTD (m), median (IQR) | −42 (−97–16) | −15 (−50–29) | 0.9 |
| IPF mPD-L1pos n = 11 | IPF mPD-L1neg n = 30 | p-Value | |
| Change FVC % (SD) | −2 ± 8 | −3.8 ± 11 | 0.61 |
| Change TLco %(SD) | −1 ± 7 | −1.6 ± 8 | 0.83 |
| Change 6MWTD (m), median (IQR) | −4 (−97–4) | −13 (−35–19) | 0.86 |
| IPF mPD-L1pos n = 11 | Non- IPF mPD-L1pos n = 4 | p-Value | |
| Change FVC % (SD) | −2 ± 8 | 0.75 ± 18 | 0.34 |
| Change TLco % (SD) | −1 ± 7 | 5 ± 13 | 0.13 |
| Change 6MWTD (m), median (IQR) | −4 (−97–4) | 1 (−121–41) | 0.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kronborg-White, S.; Madsen, L.B.; Bendstrup, E.; Poletti, V. PD-L1 Expression in Patients with Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2021, 10, 5562. https://doi.org/10.3390/jcm10235562
Kronborg-White S, Madsen LB, Bendstrup E, Poletti V. PD-L1 Expression in Patients with Idiopathic Pulmonary Fibrosis. Journal of Clinical Medicine. 2021; 10(23):5562. https://doi.org/10.3390/jcm10235562
Chicago/Turabian StyleKronborg-White, Sissel, Line Bille Madsen, Elisabeth Bendstrup, and Venerino Poletti. 2021. "PD-L1 Expression in Patients with Idiopathic Pulmonary Fibrosis" Journal of Clinical Medicine 10, no. 23: 5562. https://doi.org/10.3390/jcm10235562