
Mechanisms and Regulation of Cellular Senescence


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Senescence and the Control of Cell Cycle Arrest
2.1. Cell Cycle Exit in G1

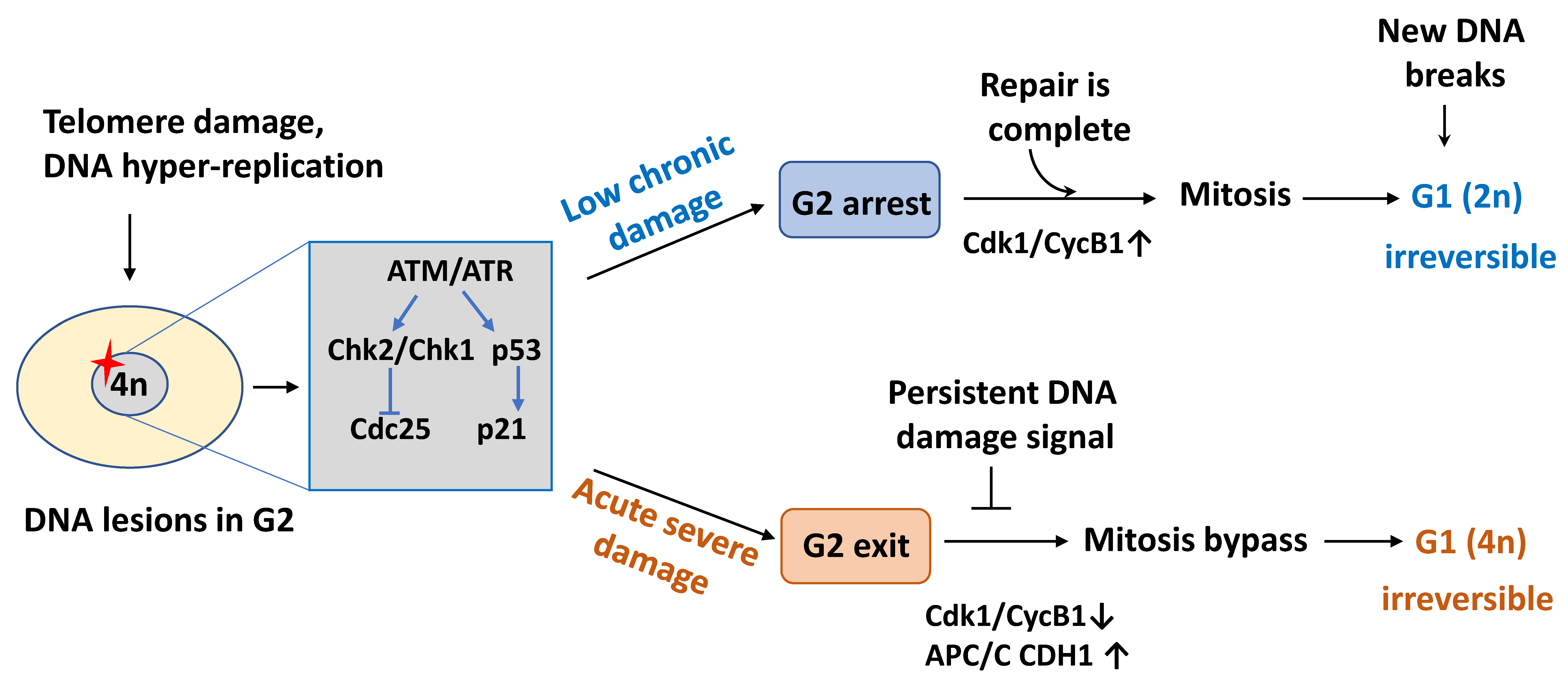
2.2. Cell Cycle Exit in G2
3. Molecular Signaling and Pathways Leading to Senescence Entry
3.1. RB/p16 and p53 Pathways Engaged upon Senescence
3.2. Short Telomeres and DNA Damage Signaling
3.3. Oxidative Damage and Irreparable Telomeric Lesions
3.4. Oncogene Activity and Replication Stress
4. Transcriptional and Post-Transcriptional Control of Senescence
4.1. The Senescence Transcriptional Program
4.1.1. Senescence Core Genes
4.1.2. Genes Implicated in the Senescence-Associated Secretory Phenotype
4.2. Post-Transcriptional Regulation of Senescence
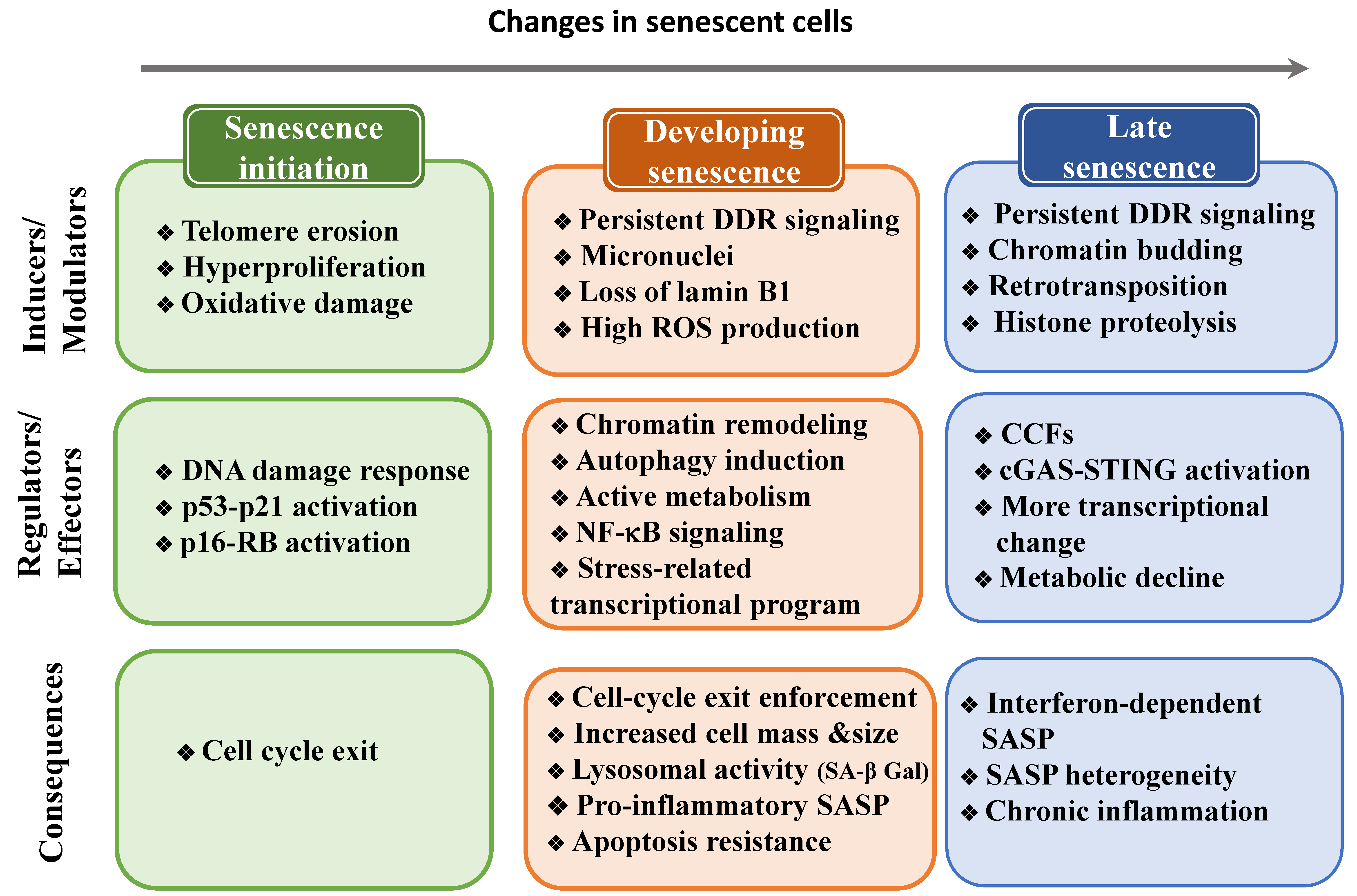
5. Changes to the Cell State during Senescence
5.1. Senescent Cell Metabolism
5.2. Cellular Structures: Membrane-Bound Organelles
5.3. Autophagy Regulation of Senescence
6. Ensuring the Duration of the Senescence State
6.1. Protection from Apoptosis during Senescence
6.2. Positive Feedback Loops
6.2.1. Mitochondrial Dysfunction and ROS Production
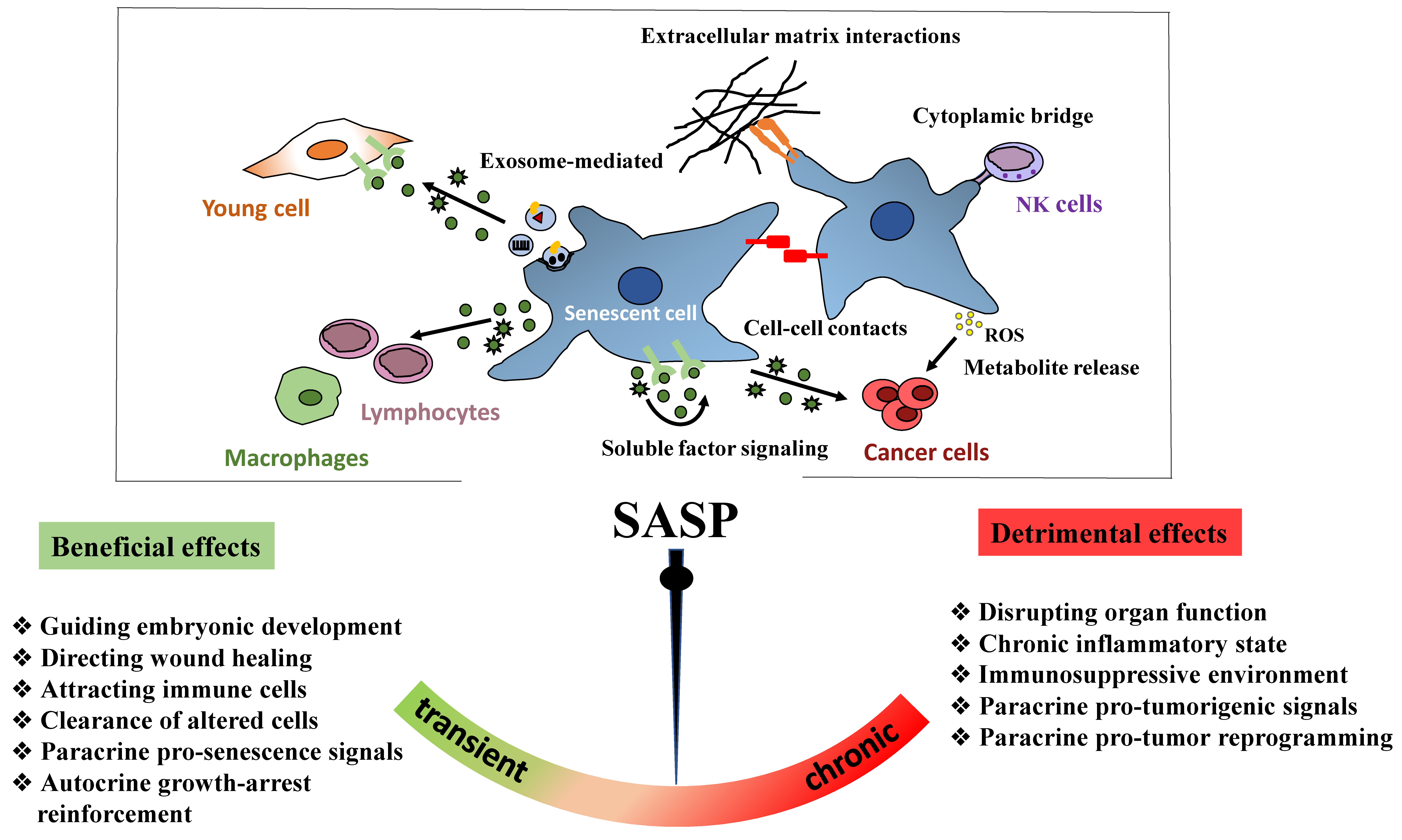
6.2.2. SASP Factors
6.3. Epigenetic Profile and Chromatin Remodeling
7. Relevance of Senescence in Cancer Development and Aging
7.1. Senescent Cells in Aging
7.1.1. Causal Role of Senescent Cells in Aging and Age-related Diseases In Vivo
7.1.2. Mechanisms by Which Senescent Cells Drive the Pathogenesis of Age-Associated Diseases
7.1.3. Inefficient Immune Clearance of Senescent Cells
7.2. Senescent Cells in Cancer
7.2.1. Cellular Senescence as a Barrier to Tumorigenesis
7.2.2. Dual Role of SASP in Cancer Pathogenesis
7.2.3. Therapy-Induced Senescence of Cancer Cells
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.I.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular Senescence in Cancer and Aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Di Fagagna, F.D. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.-P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Gire, V.; Roux, P.; Wynford-Thomas, D.; Brondello, J.-M.; Dulic, V. DNA damage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 2004, 23, 2554–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, p53, and p21CIP1, but Not p16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- De Keizer, P.L. The Fountain of Youth by Targeting Senescent Cells? Trends Mol. Med. 2017, 23, 6–17. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Weinberg, R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.-Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, S.; Coppe, J.-P.; Krtolica, A.; Campisi, J. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Z.; Jiang, H.; Jaworski, M.; Rathinam, C.; Gompf, A.; Klein, C.; Trumpp, A.; Rudolph, K.L. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat. Med. 2007, 13, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizhanovsky, V.; Yon, M.; Dickins, R.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.-I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Gire, V.; Dulić, V. Senescence from G2 arrest, revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, B.B.; Starborg, M.; Brookes, S.; Peters, G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr. Biol. 1998, 8, 351–354. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.-Y.; Campisi, J. Tumor Suppressor and Aging Biomarker p16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [Green Version]
- Noda, A.; Ning, Y.; Venable, S.F.; Pereira-Smith, O.M.; Smith, J.R. Cloning of Senescent Cell-Derived Inhibitors of DNA Synthesis Using an Expression Screen. Exp. Cell Res. 1994, 211, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [Green Version]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulicć, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Bond, J.; Haughton, M.; Blaydes, J.; Gire, V.; Wynford-Thomas, D.; Wyllie, F. Evidence That Transcriptional Activation by P53 Plays a Direct Role in the Induction of Cellular Senescence. Oncogene 1996, 13, 2097–2104. [Google Scholar]
- Brown, J.P.; Wei, W.; Sedivy, J.M. Bypass of Senescence After Disruption of p21 CIP1/WAF1 Gene in Normal Diploid Human Fibroblasts. Science 1997, 277, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.; Chen, J.-Y.; Stewart-Ornstein, J.; Karhohs, K.W.; Mock, C.S.; Lahav, G. Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 2018, 71, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, A.R.; Ju, Z.; Djojosubroto, M.W.; Schienke, A.; Lechel, A.; Schaetzlein, S.; Jiang, H.; Stepczynska, A.; Wang, C.; Buer, J.; et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat. Genet. 2007, 39, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Prigent, S.A.; Born, T.L.; Monell, C.R.; Feramisco, J.R.; Bertolaet, B.L. Microinjection of anti-p21 antibodies induces senescent Hs68 human fibroblasts to synthesize DNA but not to divide. Cancer Res. 1999, 59, 5341–5348. [Google Scholar]
- Wei, W.; Herbig, U.; Wei, S.; Dutriaux, A.; Sedivy, J.M. Loss of retinoblastoma but not p16 function allows bypass of replicative senescence in human fibroblasts. EMBOJ 2003, 4, 1061–1066. [Google Scholar] [CrossRef]
- Bond, J.; Jones, C.; Haughton, M.; DeMicco, C.; Kipling, D.; Wynford-Thomas, D. Direct evidence from siRNA-directed “knock down” that p16INK4a is required for human fibroblast senescence and for limiting ras-induced epithelial cell proliferation. Exp. Cell Res. 2004, 292, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Gire, V.; Wynford-Thomas, D. Reinitiation of DNA Synthesis and Cell Division in Senescent Human Fibroblasts by Microinjection of Anti-p53 Antibodies. Mol. Cell. Biol. 1998, 18, 1611–1621. [Google Scholar] [CrossRef] [Green Version]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Jacobs, J.J.; de Lange, T. Significant Role for p16INK4a in p53-Independent Telomere-Directed Senescence. Curr. Biol. 2004, 14, 2302–2308. [Google Scholar] [CrossRef] [Green Version]
- Bond, J.A.; Haughton, M.F.; Rowson, J.M.; Smith, P.J.; Gire, V.; Wynford-Thomas, D.; Wyllie, F.S. Control of Replicative Life Span in Human Cells: Barriers to Clonal Expansion Intermediate Between M1 Senescence and M2 Crisis. Mol. Cell. Biol. 1999, 19, 3103–3114. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Košař, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16 ink4a. Cell Cycle 2011, 10, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, S.; Rowe, J.; Ruas, M.; Llanos, S.; Clark, P.A.; Lomax, M.; James, M.C.; Vatcheva, R.; Bates, S.; Vousden, K.H.; et al. INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence. EMBO J. 2002, 21, 2936–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benanti, J.; Galloway, D.A. Normal Human Fibroblasts Are Resistant to RAS-Induced Senescence. Mol. Cell. Biol. 2004, 24, 2842–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: A role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Chandrasekarant, C.; Gordont, J.I.; Beachi, D.; Jacks, T. Radiation-Induced Cell Cycle Arrest Compromised by P21 Deficiency. Nature 1995, 377, 552–557. [Google Scholar] [CrossRef]
- Jullien, L.; Mestre, M.; Roux, P.; Gire, V. Eroded human telomeres are more prone to remain uncapped and to trigger a G2 checkpoint response. Nucleic Acids Res. 2013, 41, 900–911. [Google Scholar] [CrossRef]
- Krenning, L.; Feringa, F.; Shaltiel, I.; van den Berg, J.; Medema, R.H. Transient Activation of p53 in G2 Phase Is Sufficient to Induce Senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Johmura, Y.; Shimada, M.; Misaki, T.; Naiki-Ito, A.; Miyoshi, H.; Motoyama, N.; Ohtani, N.; Hara, E.; Nakamura, M.; Morita, A.; et al. Necessary and Sufficient Role for a Mitosis Skip in Senescence Induction. Mol. Cell 2014, 55, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baus, F.; Gire, V.; Fisher, D.; Piette, J.; Dulić, V. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J. 2003, 22, 3992–4002. [Google Scholar] [CrossRef] [Green Version]
- Charrier-Savournin, F.B.; Château, M.-T.; Gire, V.; Sedivy, J.; Piette, J.; Dulić, V. p21-Mediated Nuclear Retention of Cyclin B1-Cdk1 in Response to Genotoxic Stress. Mol. Biol. Cell 2004, 15, 3965–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müllers, E.; Cascales, H.S.; Jaiswal, H.; Saurin, A.; Lindqvist, A. Nuclear translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle 2014, 13, 2733–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müllers, E.; Cascales, H.S.; Burdova, K.; Macurek, L.; Lindqvist, A. Residual Cdk1/2 activity after DNA damage promotes senescence. Aging Cell 2017, 16, 575–584. [Google Scholar] [CrossRef]
- Zachariae, W.; Schwab, M.; Nasmyth, K.; Seufert, W. Control of Cyclin Ubiquitination by CDK-Regulated Binding of Hct1 to the Anaphase Promoting Complex. Science 1998, 282, 1721–1724. [Google Scholar] [CrossRef]
- Macedo, J.C.M.; Vaz, S.; Bakker, B.; Ribeiro, R.; Bakker, P.L.; Escandell, J.M.; Ferreira, M.G.; Medema, R.; Foijer, F.; Logarinho, E. FoxM1 repression during human aging leads to mitotic decline and aneuploidy-driven full senescence. Nat. Commun. 2018, 9, 2834. [Google Scholar] [CrossRef] [Green Version]
- Dikovskaya, D.; Cole, J.J.; Mason, S.; Nixon, C.; Karim, S.A.; McGarry, L.; Clark, W.; Hewitt, R.N.; Sammons, M.A.; Zhu, J.; et al. Mitotic Stress Is an Integral Part of the Oncogene-Induced Senescence Program that Promotes Multinucleation and Cell Cycle Arrest. Cell Rep. 2015, 12, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the Unique Role of the Retinoblastoma Tumor Suppressor during Cellular Senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA Damage Foci at Dysfunctional Telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor Suppression at the Mouse INK4a Locus Mediated by the Alternative Reading Frame Product p19 ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Sage, J.; Mulligan, G.J.; Attardi, L.D.; Miller, A.; Chen, S.; Williams, B.; Theodorou, E.; Jacks, T. Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization. Genes Dev. 2000, 14, 3037–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef]
- Sharpless, N.; Bardeesy, N.; Lee, K.-H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Hemmer, R.M.; Sedivy, J.M. Role of p14 ARF in Replicative and Induced Senescence of Human Fibroblasts. Mol. Cell. Biol. 2001, 21, 6748–6757. [Google Scholar] [CrossRef] [Green Version]
- Cipriano, R.; Kan, C.E.; Graham, J.; Danielpour, D.; Stampfer, M.; Jackson, M.W. TGF- signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8668–8673. [Google Scholar] [CrossRef] [Green Version]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.; Kieboom, K.; Marino, S.; DePinho, R.; Van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Bernard, D.; Martínez, D.; Beach, D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol. 2004, 6, 67–72. [Google Scholar] [CrossRef]
- Dietrich, N.; Bracken, A.; Trinh, E.; Schjerling, C.K.; Koseki, H.; Rappsilber, J.; Helin, K.; Hansen, K.H. Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO J. 2007, 26, 1637–1648. [Google Scholar] [CrossRef] [Green Version]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.-C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Cloos, P.A.; Rudkjær, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009, 23, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.-M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Doksani, Y.; Wu, J.Y.; de Lange, T.; Zhuang, X. Super-Resolution Fluorescence Imaging of Telomeres Reveals TRF2-Dependent T-loop Formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruis, P.; Boulton, S.J. The end protection problem—An unexpected twist in the tail. Genes Dev. 2021, 35, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Lydall, D. Hiding at the ends of yeast chromosomes: Telomeres, nucleases and checkpoint pathways. J. Cell Sci. 2003, 116, 4057–4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Schertzer, M.; Wu, X.; Gertsenstein, M.; Selig, S.; Kammori, M.; Pourvali, R.; Poon, S.; Vulto, I.; Chavez, E.; et al. Regulation of Murine Telomere Length by Rtel: An Essential Gene Encoding a Helicase-like Protein. Cell 2004, 117, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, D.M.; Rowson, J.; Wynford-Thomas, D.; Kipling, D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat. Genet. 2003, 33, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005, 7, 712–718. [Google Scholar] [CrossRef]
- Cesare, A.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Cesare, A.; Hayashi, M.T.; Crabbe, L.; Karlseder, J. The Telomere Deprotection Response Is Functionally Distinct from the Genomic DNA Damage Response. Mol. Cell 2013, 51, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Van Ly, D.; Low, R.R.J.; Frölich, S.; Bartolec, T.K.; Kafer, G.R.; Pickett, H.A.; Gaus, K.; Cesare, A.J. Telomere Loop Dynamics in Chromosome End Protection. Mol. Cell 2018, 71, 510–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdun, R.E.; Crabbe, L.; Haggblom, C.; Karlseder, J. Functional Human Telomeres Are Recognized as DNA Damage in G2 of the Cell Cycle. Mol. Cell 2005, 20, 551–561. [Google Scholar] [CrossRef]
- Stewart, S.A.; Ben-Porath, I.; Carey, V.J.; O’Connor, B.F.; Hahn, W.C.; Weinberg, R.A. Erosion of the telomeric single-strand overhang at replicative senescence. Nat. Genet. 2003, 33, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlseder, J.; Smogorzewska, A.; de Lange, T. Senescence Induced by Altered Telomere State, Not Telomere Loss. Science 2002, 295, 2446–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, Z.; Cesare, A.; Huschtscha, L.I.; Neumann, A.A.; Reddel, R.R. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2012, 13, 52–59. [Google Scholar] [CrossRef]
- Deckbar, D.; Birraux, J.; Krempler, A.; Tchouandong, L.; Beucher, A.; Walker, S.; Stiff, T.; Jeggo, P.; Loöbrich, M. Chromosome breakage after G2 checkpoint release. J. Cell Biol. 2007, 176, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Krempler, A.; Deckbar, D.; Jeggo, P.A.; Lobrich, M. An Imperfect G2M Checkpoint Contributes to Chromosome Instability Following Irradiation of S and G2Phase Cells. Cell Cycle 2007, 6, 1682–1686. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; Van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.; Passos, J.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular Senescence in Aging Primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [Green Version]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, G.; Jurk, D.; Marques, F.M.; Correia-Melo, C.; Hardy, T.L.D.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Blackburn, E.H. Telomeres and telomerase: Their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005, 579, 859–862. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, S.; Samper, E.; Krtolica, A.; Goldstein, J.; Melov, S.; Campisi, J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 2003, 5, 741–747. [Google Scholar] [CrossRef]
- De Magalhães, J.P.; Chainiaux, F.; Remacle, J.; Toussaint, O. Stress-induced premature senescence in BJ and hTERT-BJ1 human foreskin fibroblasts. FEBS Lett. 2002, 523, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Sedivy, J.M. Expression of catalytically active telomerase does not prevent premature senescence caused by overexpression of oncogenic Ha-Ras in normal human fibroblasts. Cancer Res. 1999, 59, 1539–1543. [Google Scholar] [PubMed]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions: Telomere Dysfunction Limits Cancer Progression in Humans. EMBO J. 2012, 31, 2839–2851. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broccoli, D.; Smogorzewska, A.; Chong, L.; de Lange, T. Human telomeres contain two distinct Myb–related proteins, TRF1 and TRF2. Nat. Genet. 1997, 17, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Colavitti, R.; Finkel, T. Reactive Oxygen Species as Mediators of Cellular Senescence. IUBMB Life 2005, 57, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. The power and the promise of oncogene-induced senescence markers. Nat. Rev. Cancer 2006, 6, 472–476. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.; Herlyn, M.; et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.-X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschênes-Simard, X.; Ferbeyre, G. Mitochondrial Dysfunction Contributes to Oncogene-Induced Senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyemi, U.; Lagentechevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.R.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.-M.; et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012, 31, 1117–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.Y.; Sharpless, N.E. The Regulation of INK4/ARF in Cancer and Aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta BBA-Rev. Cancer 2002, 1602, 73–87. [Google Scholar] [CrossRef]
- Ferbeyre, G.; de Stanchina, E.; Lin, A.W.; Querido, E.; McCurrach, M.E.; Hannon, G.J.; Lowe, S.W. Oncogenic ras and p53 Cooperate to Induce Cellular Senescence. Mol. Cell. Biol. 2002, 22, 3497–3508. [Google Scholar] [CrossRef] [Green Version]
- Rowland, B.D.; Denissov, S.G.; Douma, S.; Stunnenberg, H.G.; Bernards, R.; Peeper, D.S. E2F transcriptional repressor complexes are critical downstream targets of p19ARF/p53-induced proliferative arrest. Cancer Cell 2002, 2, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Munro, J.; Stott, F.J.; Vousden, K.H.; Peters, G.; Parkinson, E.K. Role of the alternative INK4A proteins in human keratinocyte senescence: Evidence for the specific inactivation of p16INK4A upon immortalization. Cancer Res. 1999, 59, 2516–2521. [Google Scholar]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Bartkova, J.; Kotsinas, A.; Pateras, I.S.; Liontos, M.; Velimezi, G.; Kosar, M.; Liloglou, T.; Trougakos, I.P.; Dyrskjøt, L.; et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 2013, 20, 1485–1497. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D.; Jones, P.A. The Human ARF Cell Cycle Regulatory Gene Promoter Is a CpG Island Which Can Be Silenced by DNA Methylation and Down-Regulated by Wild-Type p53. Mol. Cell. Biol. 1998, 18, 6457–6473. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Roussel, M.F.; Sherr, C.J. Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc. Natl. Acad. Sci. USA 1999, 96, 3993–3998. [Google Scholar] [CrossRef] [Green Version]
- Berkovich, E.; Lamed, Y.; Ginsberg, D. E2F and Ras Synergize in Transcriptionally Activating p14ARFExpression. Cell Cycle 2003, 2, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome signature of cellular senescence. Nucleic Acids Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef]
- Shelton, D.N.; Chang, E.; Whittier, P.S.; Choi, D.; Funk, W.D. Microarray analysis of replicative senescence. Curr. Biol. 1999, 9, 939–945. [Google Scholar] [CrossRef] [Green Version]
- Ly, D.H.; Lockhart, D.J.; Lerner, R.A.; Schultz, P.G. Mitotic Misregulation and Human Aging. Science 2000, 287, 2486–2492. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pan, K.-H.; Cohen, S.N. Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc. Natl. Acad. Sci. USA 2003, 100, 3251–3256. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Byun, H.; Jee, B.A.; Cho, H.; Seo, Y.; Kim, Y.; Park, M.H.; Chung, H.; Woo, H.G.; Yoon, G. Implications of time-series gene expression profiles of replicative senescence. Aging Cell 2013, 12, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Lackner, D.H.; Hayashi, M.; Cesare, A.; Karlseder, J. A genomics approach identifies senescence-specific gene expression regulation. Aging Cell 2014, 13, 946–950. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar] [PubMed]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Marcotte, R.; Lacelle, C.; Wang, E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 2004, 125, 777–783. [Google Scholar] [CrossRef]
- Seluanov, A.; Gorbunova, V.; Falcovitz, A.; Sigal, A.; Milyavsky, M.; Zurer, I.; Shohat, G.; Goldfinger, N.; Rotter, V. Change of the Death Pathway in Senescent Human Fibroblasts in Response to DNA Damage Is Caused by an Inability to Stabilize p53. Mol. Cell. Biol. 2001, 21, 1552–1564. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Hoare, M.; Ito, Y.; Kang, T.-W.; Weekes, M.P.; Matheson, N.J.; Patten, D.; Shetty, S.; Parry, A.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Malaquin, N.; Martinez, A.; Rodier, F. Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype. Exp. Gerontol. 2016, 82, 39–49. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 2011, 21, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, L.; Lu, A.; Han, Y.; Colangelo, D.; Bukata, C.; Scibetta, A.; Yousefzadeh, M.J.; Li, X.; Gurkar, A.; et al. ATM is a key driver of NF-κB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging 2020, 12, 4688–4710. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype: P38 Regulates the Senescence Secretory Phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; DeMaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- González, A.; Hall, M.N.; Lin, S.-C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA Damage Signaling Triggers Degradation of Histone Methyltransferases through APC/CCdh1 in Senescent Cells. Mol. Cell 2012, 45, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Leon, K.E.; Buj, R.; Lesko, E.; Dahl, E.S.; Chen, C.-W.; Tangudu, N.K.; Imamura-Kawasawa, Y.; Kossenkov, A.V.; Hobbs, R.P.; Aird, K.M. DOT1L modulates the senescence-associated secretory phenotype through epigenetic regulation of IL1A. J. Cell Biol. 2021, 220, e202008101. [Google Scholar] [CrossRef]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.; et al. Formation of MacroH2A-Containing Senescence-Associated Heterochromatin Foci and Senescence Driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Tanizawa, H.; Fatkhutdinov, N.; Bitler, B.; Le, L.; Alicea, G.; Yang, T.-L.; Johnson, F.B.; et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J. Cell Biol. 2016, 215, 325–334. [Google Scholar] [CrossRef]
- Tasdemir, N.; Banito, A.; Roe, J.-S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.-H.; Aksoy, O.; Bolden, J.E.; Chen, C.-C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef]
- Harding, S.; Benci, J.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [Green Version]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Simon, M.; Van Meter, M.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; De Cecco, M.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 2019, 29, 871–885. [Google Scholar] [CrossRef] [Green Version]
- Masuda, K.; Kuwano, Y.; Nishida, K.; Rokutan, K. General RBP expression in human tissues as a function of age. Ageing Res. Rev. 2012, 11, 423–431. [Google Scholar] [CrossRef]
- Wang, W. Regulatory RNA-binding proteins in senescence. Ageing Res. Rev. 2012, 11, 485–490. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Srikantan, S.; Kuwano, Y.; Gorospe, M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc. Natl. Acad. Sci. USA 2008, 105, 20297–20302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Sanduja, S.; Kaza, V.; Dixon, D.A. The mRNA decay factor tristetraprolin (TTP) induces senescence in human papillomavirus-transformed cervical cancer cells by targeting E6-AP ubiquitin ligase. Aging 2009, 1, 803–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Xu, G.; Ghandhi, S.; Hubbard, K. Modulation of the expression of p16INK4a and p14ARF by hnRNP A1 and A2 RNA binding proteins: Implications for cellular senescence. J. Cell. Physiol. 2002, 193, 19–25. [Google Scholar] [CrossRef]
- Wang, W.; Martindale, J.L.; Yang, X.; Chrest, F.J.; Gorospe, M. Increased stability of the p16 mRNA with replicative senescence. EMBO Rep. 2005, 6, 158–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga-Yamanaka, K.; Abdelmohsen, K.; Martindale, J.L.; Yang, X.; Taub, D.D.; Gorospe, M. NF90 coordinately represses the senescence-associated secretory phenotype. Aging 2012, 4, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Dutertre, M.; Lambert, S.; Carreira, A.; Amor-Guéret, M.; Vagner, S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem. Sci. 2014, 39, 141–149. [Google Scholar] [CrossRef]
- Ohsawa, R.; Seol, J.-H.; Tyler, J.K. At the intersection of non-coding transcription, DNA repair, chromatin structure, and cellular senescence. Front. Genet. 2013, 4, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009, 21, 452–460. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Bushati, N.; Cohen, S.M. microRNA Functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Gorospe, M. Noncoding RNA control of cellular senescence: Senescence NcRNAs. Wiley Interdiscip. Rev. RNA 2015, 6, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zheng, Q.; Zhao, G.; Yuan, W.; Liu, W. Regulation of cellular senescence by microRNAs. Mech. Ageing Dev. 2020, 189, 111264. [Google Scholar] [CrossRef] [PubMed]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Deschênes, M.; Chabot, B. The emerging role of alternative splicing in senescence and aging. Aging Cell 2017, 16, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, S.A.; Grochova, D.; McKenna, T.; Borate, B.; Trivedi, N.S.; Erdos, M.R.; Eriksson, M. Global genome splicing analysis reveals an increased number of alternatively spliced genes with aging. Aging Cell 2016, 15, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, Z.; Ma, T.; Wei, G.; Ni, T. Alternative splicing in aging and age-related diseases. Transl. Med. Aging 2017, 1, 32–40. [Google Scholar] [CrossRef]
- Lin, C.-L.G.; Bristol, L.A.; Jin, L.; Dykes-Hoberg, M.; Crawford, T.; Clawson, L.; Rothstein, J.D. Aberrant RNA Processing in a Neurodegenerative Disease: The Cause for Absent EAAT2, a Glutamate Transporter, in Amyotrophic Lateral Sclerosis. Neuron 1998, 20, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-Dependent Nuclear Defects in Human Aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Blanco, F.-J.; Grande, M.T.; Langa, C.; Oujo, B.; Velasco, S.; Rodriguez-Barbero, A.; Pérez-Gómez, E.; Quintanilla, M.; Loópez-Novoa, J.M.; Bernabéu, C. S-Endoglin Expression Is Induced in Senescent Endothelial Cells and Contributes to Vascular Pathology. Circ. Res. 2008, 103, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.-M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Crutchley, J.; Zhang, D.; Owzar, K.; Kastan, M.B. Identification of a DNA Damage–Induced Alternative Splicing Pathway That Regulates p53 and Cellular Senescence Markers. Cancer Discov. 2017, 7, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Georgilis, A.; Klotz, S.; Hanley, C.J.; Herranz, N.; Weirich, B.; Morancho, B.; Leote, A.C.; D’Artista, L.; Gallage, S.; Seehawer, M.; et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell 2018, 34, 85–102. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells—Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Goldstein, S.; Ballantyne, S.R.; Robson, A.L.; Moerman, E.J. Energy metabolism in cultured human fibroblasts during aging in vitro. J. Cell. Physiol. 1982, 112, 419–424. [Google Scholar] [CrossRef]
- Zwerschke, W.; Mazurek, S.; Stöckl, P.; Hütter, E.; Eigenbrodt, E.; Jansen-Dürr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 2003, 376, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.; Von Zglinicki, T. Mitochondrial dysfunction and cell senescence—Skin deep into mammalian aging. Aging 2012, 4, 74–75. [Google Scholar] [CrossRef]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.; Mackay, G.; Van Der Burg, S.H.; Verdegaal, E.M.E.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef]
- James, E.; Michalek, R.D.; Pitiyage, G.N.; De Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent Human Fibroblasts Show Increased Glycolysis and Redox Homeostasis with Extracellular Metabolomes That Overlap with Those of Irreparable DNA Damage, Aging, and Disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef]
- Lu, T.; Finkel, T. Free radicals and senescence. Exp. Cell Res. 2009, 314, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Wenz, T. Mitochondria and PGC-1α in Aging and Age-Associated Diseases. J. Aging Res. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta BBA-Gen. Subj. 2008, 1780, 1291–1303. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.M.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Quijano, C.; Cao, L.; Fergusson, M.M.; Romero, H.; Liu, J.; Gutkind, S.; Rovira, I.I.; Mohney, R.P.; Karoly, E.D.; Finkel, T. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 2012, 11, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Durbin, K.R.; Sweet, S.M.M.; Tipton, J.D.; Zheng, Y.; Kelleher, N.L. Oncogene-induced cellular senescence elicits an anti-Warburg effect. Proteomics 2013, 13, 2585–2596. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, R. Metabolic alterations accompanying oncogene-induced senescence. Mol. Cell. Oncol. 2014, 1, e963481. [Google Scholar] [CrossRef] [Green Version]
- Aird, K.M.; Zhang, R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett. 2015, 356, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Aird, K.M.; Worth, A.J.; Snyder, N.; Lee, J.V.; Sivanand, S.; Liu, Q.; Blair, I.A.; Wellen, K.E.; Zhang, R. ATM Couples Replication Stress and Metabolic Reprogramming during Cellular Senescence. Cell Rep. 2015, 11, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Burkewitz, K.; Zhang, Y.; Mair, W.B. AMPK at the Nexus of Energetics and Aging. Cell Metab. 2014, 20, 10–25. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-Activated Protein Kinase Induces a p53-Dependent Metabolic Checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, X.; López de Silanes, I.; Carling, D.; Gorospe, M. Increased AMP:ATP Ratio and AMP-activated Protein Kinase Activity during Cellular Senescence Linked to Reduced HuR Function. J. Biol. Chem. 2003, 278, 27016–27023. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Puzio-Kuter, A.M. The Role of p53 in Metabolic Regulation. Genes Cancer 2011, 2, 385–391. [Google Scholar] [CrossRef]
- Takebayashi, S.; Tanaka, H.; Hino, S.; Nakatsu, Y.; Igata, T.; Sakamoto, A.; Narita, M.; Nakao, M. Retinoblastoma protein promotes oxidative phosphorylation through upregulation of glycolytic genes in oncogene-induced senescent cells. Aging Cell 2015, 14, 689–697. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Teresky, A.K.; Hernando, E.; Cordon-Cardo, C.; Levine, A.J. Declining p53 function in the aging process: A possible mechanism for the increased tumor incidence in older populations. Proc. Natl. Acad. Sci. USA 2007, 104, 16633–16638. [Google Scholar] [CrossRef] [Green Version]
- Tonnessen-Murray, C.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-S.; Hinds, P.W. Increased Ezrin Expression and Activation by CDK5 Coincident with Acquisition of the Senescent Phenotype. Mol. Cell 2003, 14, 1163–1176. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-Associated β-Galactosidase Reflects an Increase in Lysosomal Mass during Replicative Ageing of Human Endothelial Cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Severino, J.; Allen, R.G.; Balin, S.; Balin, A.; Cristofalo, V.J. Is β-Galactosidase Staining a Marker of Senescence in Vitro and in Vivo? Exp. Cell Res. 2000, 257, 162–171. [Google Scholar] [CrossRef]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez, M.P.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2012, 5, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Ferry, C.H.; Markell, L.K.; Blazanin, N.; Glick, A.B.; Gonzalez, F.J.; Peters, J.M. The Nuclear Receptor Peroxisome Proliferator-activated Receptor-β/δ (PPARβ/δ) Promotes Oncogene-induced Cellular Senescence through Repression of Endoplasmic Reticulum Stress. J. Biol. Chem. 2014, 289, 20102–20119. [Google Scholar] [CrossRef] [Green Version]
- Matos, L.; Gouveia, A.; Almeida, H. ER Stress Response in Human Cellular Models of Senescence. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2015, 70, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baraibar, M.A.; Liu, L.; Ahmed, E.; Friguet, B. Protein Oxidative Damage at the Crossroads of Cellular Senescence, Aging, and Age-Related Diseases. Oxidative Med. Cell. Longev. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 Activated by Proteolysis Binds in the Presence of NF-Y (CBF) Directly to the cis -Acting Element Responsible for the Mammalian Unfolded Protein Response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [Green Version]
- Geiss-Friedlander, R.; Jarosch, E.; Urban, J.; Volkwein, C.; Sommer, T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2000, 2, 379–384. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanzo, R.; Bartkova, J.; Merchut-Maya, J.M.; Hall, A.; Bouchal, J.; Dyrskjøt, L.; Frankel, L.B.; Gorgoulis, V.; Maya-Mendoza, A.; Jäättelä, M.; et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2020, 27, 1134–1153. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.J.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial Coupling of mTOR and Autophagy Augments Secretory Phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [Green Version]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.-A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Chen, H.; Gong, H.; Tang, X.; Huang, N.; Xu, W.; Tai, H.; Zhang, G.; Zhao, T.; Gong, C.; et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress–induced senescence. J. Biol. Chem. 2020, 295, 4451–4463. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 5, 4172. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Liu, J.; Merrett, J.B. Apoptosis or Senescence-like Growth Arrest: Influence of Cell-Cycle Position, P53, P21 and Bax in H2O2 Response of Normal Human FIbroblasts. Biochem. J. 2000, 347, 543–551. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone Modifications in Senescence-Associated Resistance to Apoptosis by Oxidative Stress. Redox Biol. 2013, 1, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar] [PubMed]
- Xia, M.; Knezevic, D.; Vassilev, L.T. p21 does not protect cancer cells from apoptosis induced by nongenotoxic p53 activation. Oncogene 2011, 30, 346–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catz, S.D.; Johnson, J.L. Transcriptional Regulation of Bcl-2 by Nuclear Factor KB and Its Signicance in Prostate Cancer. Oncogene 2001, 20, 7342–7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; Zuffo, R.D.; Matti, V.; D’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Yun, M.H.; Davaapil, H.; Brockes, J. Recurrent turnover of senescent cells during regeneration of a complex structure. eLife 2015, 4, e05505. [Google Scholar] [CrossRef] [PubMed]
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells—Biological significance in cancer- and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [CrossRef] [Green Version]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saretzki, G.; Murphy, M.P.; Von Zglinicki, T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2003, 2, 141–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.-I.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Passos, J.F. Demystifying the role of mitochondria in senescence. Mol. Cell. Oncol. 2016, 3, e1162896. [Google Scholar] [CrossRef] [Green Version]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of Interferon-a/b Signalling to P53 Responses in Tumour Suppression and Antiviral Defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, O.; Mallette, F.A.; Mukhopadhyay, U.K.; Moores, A.; Ferbeyre, G. DNA Damage Signaling and P53-Dependent Senescence after Prolonged b-Interferon Stimulation. Mol. Biol. Cell 2006, 17, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular Dissection of Formation of Senescence-Associated Heterochromatin Foci. Mol. Cell. Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nuñez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A Novel Role for High-Mobility Group A Proteins in Cellular Senescence and Heterochromatin Formation. Cell 2006, 126, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, T.; Kirschner, K.; Thuret, J.-Y.; Pope, B.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.; Salama, R.; Carroll, T.; et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.; Narita, M.; Pérez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013, 27, 1800–1808. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Swanson, E.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013, 203, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; VanderKraats, N.D.; Shah, P.P.; Van Tuyn, J.; Rai, T.S.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef]
- De Cecco, M.; Criscione, S.W.; Peterson, A.L.; Neretti, N.; Sedivy, J.M.; Kreiling, J.A. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging 2013, 5, 867–883. [Google Scholar] [CrossRef] [Green Version]
- Tasselli, L.; Xi, Y.; Zheng, W.; Tennen, R.I.; Odrowaz, Z.; Simeoni, F.; Li, W.; Chua, K.F. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 2016, 23, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.; Neretti, N.; Sedivy, J.M. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef]
- Chan, A.S.L.; Narita, M. Short-term gain, long-term pain: The senescence life cycle and cancer. Genes Dev. 2019, 33, 127–143. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prata, L.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2020, 21, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Ramsey, M.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Gregory, M.H.; Tchkonia, T.; Lebrasseur, N.K.; Kirkland, J.L.; Kritchevsky, S.B.; Nicklas, B.J. Cellular Senescence Biomarker p16INK4a+ Cell Burden in Thigh Adipose is Associated with Poor Physical Function in Older Women. J. Gerontol. Ser. A 2018, 73, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; Van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, M.A.; Lee, H.-W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.; Greider, C.W. Telomere Shortening and Tumor Formation by Mouse Cells Lacking Telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Jaskelioff, M.; Muller, F.L.; Paik, J.-H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadiñanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-W.; Blasco, M.A.; Gottlieb, G.J.; Horner, J.W.; Greider, C.W.; DePinho, R. Essential role of mouse telomerase in highly proliferative organs. Nature 1998, 392, 569–574. [Google Scholar] [CrossRef]
- Rudolph, K.L.; Chang, S.; Lee, H.-W.; Blasco, M.A.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Multani, A.S.; Cabrera, N.G.; Naylor, M.L.; Laud, P.; Lombard, D.; Pathak, S.; Guarente, L.; DePinho, R. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 2004, 36, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; DePinho, R.A. Inhibition of Experimental Liver Cirrhosis in Mice by Telomerase Gene Delivery. Science 2000, 287, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; Hu, B.; Mao, X.; Rashid, A.; Li, J.; Li, J.; Liao, W.-T.; Whitley, E.M.; Dey, P.; Hou, P.; et al. Telomere dysfunction activates YAP1 to drive tissue inflammation. Nat. Commun. 2020, 11, 4766. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R. A Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395. [Google Scholar] [CrossRef]
- Bär, C.; Blasco, M.A. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Research 2016, 5, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniali, L.; Benetos, A.; Susser, E.; Kark, J.D.; Labat, C.; Kimura, M.; Desai, K.K.; Granick, M.; Aviv, A. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat. Commun. 2013, 4, 1597. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of telomere length across human tissues. Science 2020, 369, eaaz6876. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; Von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice: Senescent Cells in Aging Mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fielder, E.; von Zglinicki, T.; Jurk, D. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J. Alzheimer’s Dis. 2017, 60, S107–S131. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Matjusaitis, M.; Chin, G.; Sarnoski, E.A.; Stolzing, A. Biomarkers to identify and isolate senescent cells. Ageing Res. Rev. 2016, 29, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; DeMaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; DePinho, R.; Sharpless, N.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006, 443, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa-Victor, P.; Gutarra, S.; García-Prat, L.; Rodriguez-Ubreva, J.; Ortet-Cortada, L.; Ruiz-Bonilla, V.; Jardí, M.; Ballestar, E.; González, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Palmer, A.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 2015, 4, e12997. [Google Scholar] [CrossRef]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.-M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Grants, J.M.; Wegrzyn, J.; Hui, T.; O’Neill, K.; Shadbolt, M.; Knapp, D.J.H.F.; Parker, J.; Deng, Y.; Gopal, A.; Docking, T.R.; et al. Altered microRNA expression links IL6 and TNF-induced inflammaging with myeloid malignancy in humans and mice. Blood 2020, 135, 2235–2251. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, P.; Ogrodnik, M.; Kucheryavenko, O.; Glibert, J.; Miwa, S.; Cameron, K.; Ishaq, A.; Saretzki, G.; Nagaraja-Grellscheid, S.; Nelson, G.; et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 2019, 18, e12848. [Google Scholar] [CrossRef]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘Bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.-H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Kohli, J.; Veenstra, I.; Demaria, M. The struggle of a good friend getting old: Cellular senescence in viral responses and therapy. EMBO Rep. 2021, 22, e52243. [Google Scholar] [CrossRef]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.G.P.L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef] [PubMed]
- Wissler Gerdes, E.O.; Vanichkachorn, G.; Verdoorn, B.P.; Hanson, G.J.; Joshi, A.Y.; Murad, M.H.; Rizza, S.A.; Hurt, R.T.; Tchkonia, T.; Kirkland, J.L. Role of Senescence in the Chronic Health Consequences of COVID-19. Transl. Res. 2021, S1931524421002590. [Google Scholar] [CrossRef]
- Sagiv, A.; Burton, D.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, O.N.L.; Rodier, F.; Fontaine, F.; Coppe, J.-P.; Campisi, J.; DeGregori, J.; Laverdière, C.; Kokta, V.; Haddad, E.; Beauséjour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status: IR Exposure and Aging Markers. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef] [Green Version]
- DeMaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanoff, H.K.; Deal, A.M.; Krishnamurthy, J.; Torrice, C.; Dillon, P.; Sorrentino, J.; Ibrahim, J.G.; Jolly, T.A.; Williams, G.; Carey, L.A.; et al. Effect of Cytotoxic Chemotherapy on Markers of Molecular Age in Patients with Breast Cancer. JNCI J. Natl. Cancer Inst. 2014, 106, dju057. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.I.; Devine, O.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 2019, 10, 2387. [Google Scholar] [CrossRef] [PubMed]
- Artandi, S.E.; DePinho, R. Telomeres and telomerase in cancer. Carcinogenesis 2010, 31, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Meeker, A.K.; Hicks, J.L.; Iacobuzio-Donahue, C.A.; Montgomery, E.A.; Westra, W.H.; Chan, T.Y.; Ronnett, B.M.; De Marzo, A.M. Telomere Length Abnormalities Occur Early in the Initiation of Epithelial Carcinogenesis. Clin. Cancer Res. 2004, 10, 3317–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, L.; Jones, R.E.; Heppel, N.H.; Williams, G.T.; Sampson, J.R.; Baird, D.M. Extensive Telomere Erosion in the Initiation of Colorectal Adenomas and Its Association with Chromosomal Instability. JNCI J. Natl. Cancer Inst. 2013, 105, 1202–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, J.W.; Wright, W.E.; Werbin, H. Defining the molecular mechanisms of human cell immortalization. Biochim. Biophys. Acta BBA-Rev. Cancer 1991, 1072, 1–7. [Google Scholar] [CrossRef]
- Maser, R.S.; DePinho, R.A. Connecting Chromosomes, Crisis, and Cancer. Science 2002, 297, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Cesare, A.; Rivera, T.; Karlseder, J. Cell death during crisis is mediated by mitotic telomere deprotection. Nature 2015, 522, 492–496. [Google Scholar] [CrossRef] [Green Version]
- Artandi, S.E.; Chang, S.; Lee, S.-L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Oh, S.; Grimstead, J.W.; Zimbric, J.; Roger, L.; Heppel, N.H.; Ashelford, K.; Liddiard, K.; Hendrickson, E.A.; Baird, D.M. Escape from Telomere-Driven Crisis Is DNA Ligase III Dependent. Cell Rep. 2014, 8, 1063–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, K.; de Solórzano, C.O.; Knowles, D.; Jones, A.; Chou, W.; Rodriguez, E.G.; Kuo, W.-L.; Ljung, B.-M.; Chew, K.; Myambo, K.; et al. In situ analyses of genome instability in breast cancer. Nat. Genet. 2004, 36, 984–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.T.; Letsolo, B.T.; Jones, R.E.; Rowson, J.; Pratt, G.; Hewamana, S.; Fegan, C.; Pepper, C.; Baird, D.M. Telomere dysfunction and fusion during the progression of chronic lymphocytic leukemia: Evidence for a telomere crisis. Blood 2010, 116, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Abe, S.; Huda, N.; Tu, L.; Beam, M.J.; Grimes, B.; Gilley, D. Telomere fusions in early human breast carcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 14098–14103. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Sagiv, A.; Biran, A.; Yon, M.; Simon, J.D.; Lowe, S.W.; Krizhanovsky, V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 2012, 32, 1971–1977. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Hornsby, P.J. Senescent Human Fibroblasts Increase the Early Growth of Xenograft Tumors via Matrix Metalloproteinase Secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. JNCI J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a PARP-1 and nuclear factor-κB-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin. Cancer Biol. 2020, 65, 13–27. [Google Scholar] [CrossRef]
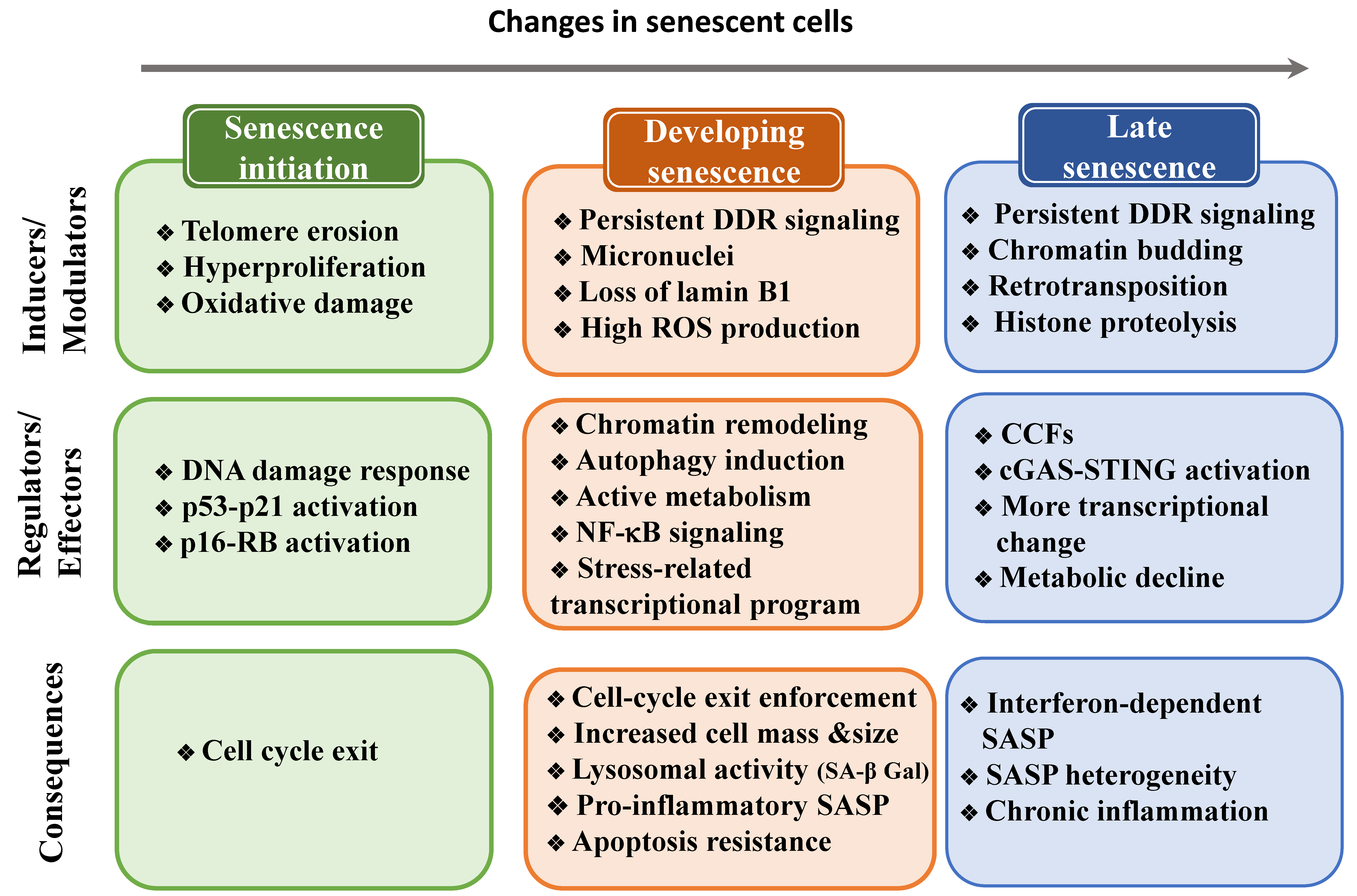
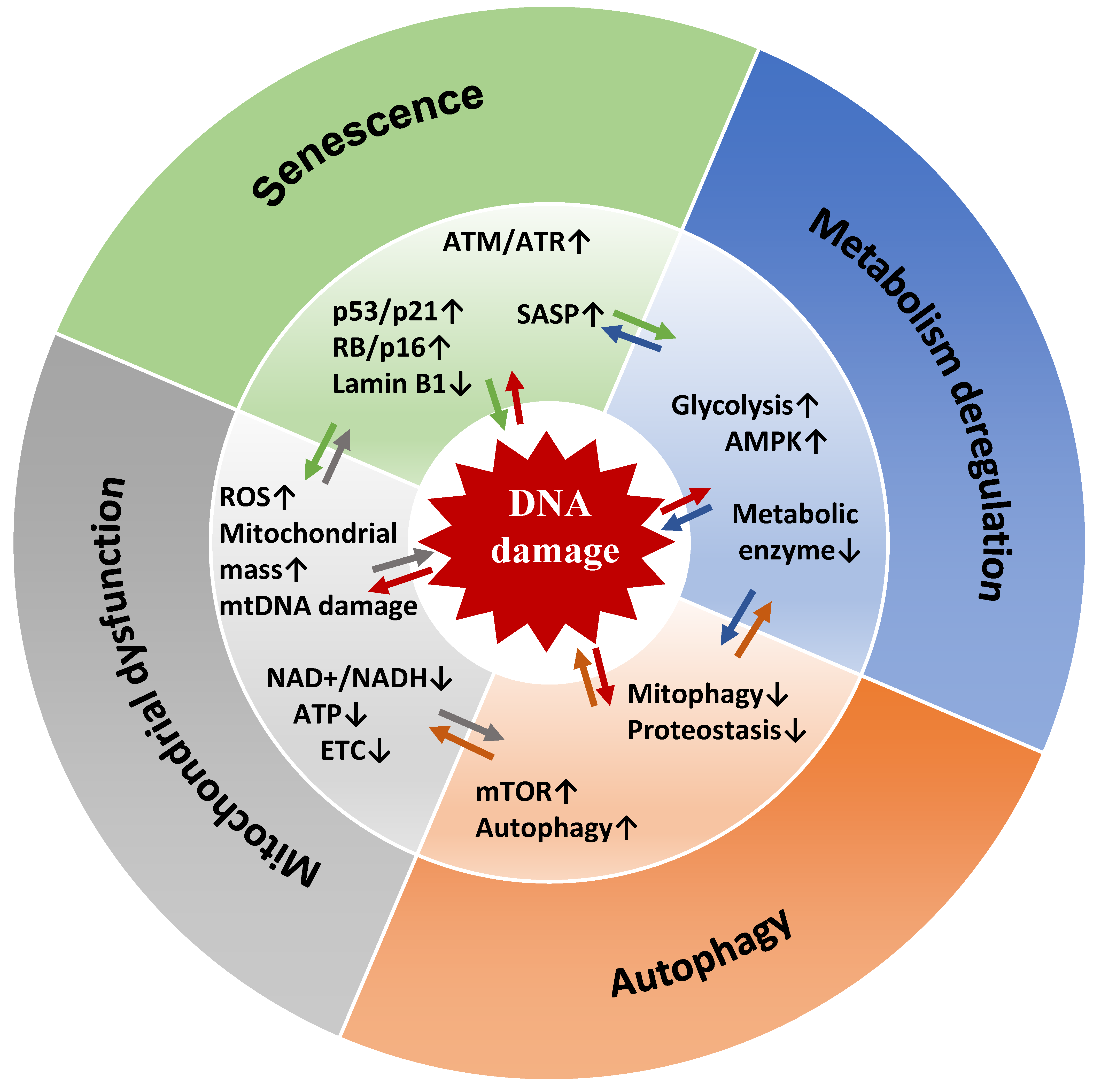
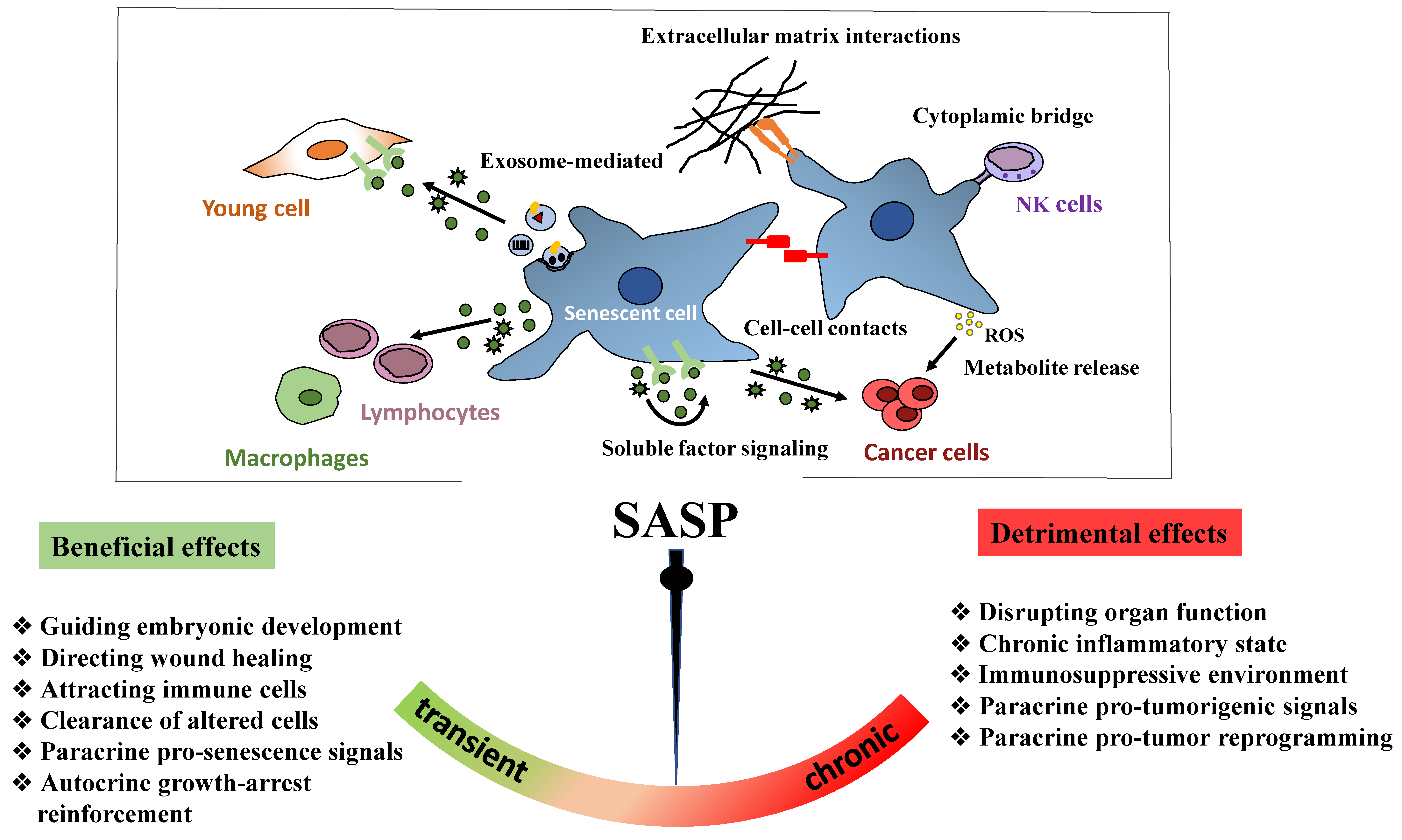
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. https://doi.org/10.3390/ijms222313173
Roger L, Tomas F, Gire V. Mechanisms and Regulation of Cellular Senescence. International Journal of Molecular Sciences. 2021; 22(23):13173. https://doi.org/10.3390/ijms222313173
Chicago/Turabian StyleRoger, Lauréline, Fanny Tomas, and Véronique Gire. 2021. "Mechanisms and Regulation of Cellular Senescence" International Journal of Molecular Sciences 22, no. 23: 13173. https://doi.org/10.3390/ijms222313173
APA StyleRoger, L., Tomas, F., & Gire, V. (2021). Mechanisms and Regulation of Cellular Senescence. International Journal of Molecular Sciences, 22(23), 13173. https://doi.org/10.3390/ijms222313173