Abstract
C-reactive protein (CRP) is well-known as a sensitive albeit unspecific biomarker of inflammation. In most rheumatic conditions, the level of this evolutionarily highly conserved pattern recognition molecule conveys reliable information regarding the degree of ongoing inflammation, driven mainly by interleukin-6. However, the underlying causes of increased CRP levels are numerous, including both infections and malignancies. In addition, low to moderate increases in CRP predict subsequent cardiovascular events, often occurring years later, in patients with angina and in healthy individuals. However, autoimmune diseases characterized by the Type I interferon gene signature (e.g., systemic lupus erythematosus, primary Sjögren’s syndrome and inflammatory myopathies) represent exceptions to the general rule that the concentrations of CRP correlate with the extent and severity of inflammation. In fact, adequate levels of CRP can be beneficial in autoimmune conditions, in that they contribute to efficient clearance of cell remnants and immune complexes through complement activation/modulation, opsonization and phagocytosis. Furthermore, emerging data indicate that CRP constitutes an autoantigen in systemic lupus erythematosus. At the same time, the increased risks of cardiovascular and cerebrovascular diseases in patients diagnosed with systemic lupus erythematosus and rheumatoid arthritis are well-established, with significant impacts on quality of life, accrual of organ damage, and premature mortality. This review describes CRP-mediated biological effects and the regulation of CRP release in relation to aspects of cardiovascular disease and mechanisms of autoimmunity, with particular focus on systemic lupus erythematosus.
1. Introduction
Although more than 90 years have passed since the discovery of C-reactive protein (CRP) at The Rockefeller University, our current understanding of CRP is essentially based on the original observations made by William S. Tillett and Thomas Francis Jr. in the laboratory of Oswald T. Avery. They found that sera obtained from patients during the acute phase of pneumococcal pneumonia precipitated with the C-polysaccharide derived from the cell wall of the pneumococcus, and that this reaction diminished as the patients recovered [1,2]. This previously unknown C-reactive substance was later found to be a protein, and thus was named “C-reactive protein” [1,2]. The ligand to which CRP bound associated with teichoic acid and was identified in the 1970s as phosphorylcholine, which is abundant on the surfaces of microbes and apoptotic cells [3].
Today, we know that CRP is a highly conserved and ubiquitous protein in vertebrates and invertebrates [4]. In humans, CRP is a liver-derived acute-phase protein that consists of five identical 23-kDa globular subunits arranged in a pentameric structure with a discoid shape. In addition to the short pentraxins, CRP and serum amyloid P component (SAP), the pentraxin superfamily contains long pentraxins, i.e., neuronal pentraxin 1 (NPTX1), neuronal pentraxin 2 (NPTX2), neuronal pentraxin receptor (NPTXR), pentraxin 3 (PTX3), and pentraxin 4 (PTX4) [5].
The integrity of the native pentameric structure of CRP (pCRP) is dependent upon the presence of calcium ions. This structure is disrupted irreversibly into monomers under denaturing conditions, e.g., in an acidic microenvironment. Such CRP monomers (mCRP) appear to have distinct biological properties, which are often different from those of pCRP [6]. In addition, mCRP has been shown to act as an autoantigen in systemic lupus erythematosus (SLE), as well as in certain other diseases [7].
CRP is produced in large quantities by hepatocytes, mainly in response to the pro-inflammatory cytokine interleukin-6 (IL-6) [8]. The profound clinical interest in CRP arises from its use as a sensitive biomarker of ongoing bacterial infections, trauma, ischemic cardiovascular disease (CVD) and other inflammatory conditions, as well as its use as a crude discriminator of bacterial from viral infections, since bacterial infections typically yield higher levels of circulating CRP. However, in conditions characterized by the Type I interferon (IFN) gene signature (e.g., SLE, primary Sjögren’s syndrome and inflammatory myopathies), CRP appears to be an unreliable marker of inflammation, since the circulating levels of CRP can be modest—despite the presence of extensive inflammation, as evidenced by an increased level of IL-6 in the circulation [9,10]. Furthermore, several studies of cardiovascular and autoimmune diseases have highlighted the importance of the genetic regulation of CRP [11,12].
In parallel with the discovery that a low-level increase in CRP is a useful risk marker for cardiovascular events, substantial progress has been made over the last decades concerning the biological properties and physiological importance of CRP in both health and disease. This review summarizes recent discoveries related to CRP-mediated biological effects, as well as to the regulation of CRP release with respect to aspects of CVD and mechanisms of autoimmunity.
2. CRP as a Biomarker in Rheumatologic Diseases
CRP is the main biomarker of inflammation used in modern healthcare. In most laboratories in Europe, for routine detection of CRP, the cut-off defining an abnormal level is set at 5 or 10 mg/L. However, for estimation of CVD risk, a ‘high-sensitivity’ CRP assay is usually applied [13,14]. At Linköping University Hospital (Sweden), the lower limit of quantification for this high-sensitivity CRP assay is 0.15 mg/L.
Historically, CRP has not always been the most popular biomarker reflecting inflammation. Several other acute-phase proteins show different concentration pattern changes in the plasma over time; some of these increase (e.g., serum amyloid A) and some decrease (e.g., albumin) during the acute-phase response [15,16]. In rheumatology, the erythrocyte sedimentation rate (ESR), which is a reflector of ongoing inflammation, deserves special attention. However, whereas the kinetics of ESR is slightly different from that of CRP, it conveys different information and can be affected by various factors, such as the erythrocyte count and fibrinogen and immunoglobulin concentrations.
In the newest set of classification criteria for rheumatoid arthritis (RA), ‘abnormal CRP and/or ESR’ is regarded as a separate item together with joint involvement, presence of autoantibodies and duration of symptoms [17]. CRP levels >10 mg/L are frequently seen in untreated patients with recent-onset RA. Other types of arthritis show different tendencies to display abnormal CRP levels. During an attack of gout, the concentration of CRP can become impressively high, often arousing a suspicion of septic arthritis. In spondylo-arthritides, such as psoriatic arthritis (PsoA), high CRP levels are usually less common, although patients with involvement of large joints may constitute exceptions to this. Consequently, abnormal levels of acute-phase proteins were not included in the classification criteria for PsoA [18].
In giant cell arteritis (GCA), unexplained high levels of CRP and ESR, accompanied by unspecific symptoms such as weight-loss and headache, may lead to a correct diagnosis [19]. GCA may present with or without proximal muscular pain, referred to as polymyalgia rheumatica (PMR). Besides muscular involvement, the 2012 classification criteria for PMR require both age ≥50 years and abnormal CRP and/or ESR levels [20]. In cases of anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitis, high levels of CRP elevation are almost ubiquitous and appear to be associated with a higher risk of renal involvement [21].
While CRP levels usually parallel disease activity in inflammatory states, it is widely accepted that CRP is an unreliable biomarker in active SLE. Still, substantial CRP responses are observed in subsets of patients with SLE with certain manifestations (e.g., serositis and polyarthritis) [10,22]. In similarity to trivial viral infections, wherein the CRP levels typically remain low, SLE may manifest as oral ulcers, pleuritis/pericarditis and leukopenia, all of which commonly affect patients with viral infections. Another feature shared by viral infections and systemic inflammatory conditions, such as SLE, primary Sjögren’s syndrome and inflammatory myopathies, is the activation of the Type I interferon system [23,24]. This will be discussed in depth below (Section 5).
Although CRP is a valuable biomarker in the clinical management of several rheumatic conditions, it must always be interpreted with caution and in the context of the symptoms presented by the patient. Several of the immunosuppressive agents used in rheumatology render the patients more prone to infections; this is particularly true for high doses of corticosteroids [25]. The risks for malignancies and paraneoplastic syndromes, which mimic rheumatic diseases, are important to consider, especially as the risks for certain cancers are increased in patients with rheumatic diseases [26,27]. Finally, some of the immunosuppressive drugs in use today directly affect the ability of the hepatocytes to produce adequate levels of CRP. The most obvious examples of this are the IL-6 receptor inhibitors tocilizumab and sarilumab, which are mainly used in cases of RA, systemic juvenile idiopathic arthritis and GCA [28]. Moreover, IL-6 signaling (and consequently CRP) may also be significantly negatively affected in patients who are receiving Janus kinase inhibitors and high doses of corticosteroids.
3. CRP as a Biomarker Indicating Increased Risks of Cerebrovascular and Cardiovascular Diseases
Based on the results of several prospective epidemiologic studies, CRP has emerged as one of the most powerful predictors of CVD in the general population [29]. In the ‘Fragmin during Instability in Coronary Artery Disease’ (FRISC) trial, which included almost 1000 patients with unstable coronary artery disease, the CRP levels were strongly associated with long-term risk of death from cardiac causes, independently of other established risk factors (i.e., hypertension, smoking, diabetes, dyslipidemia) [14]. Furthermore, CRP has been shown to contribute to several stages of atherogenesis, such as endothelial dysfunction, atherosclerotic plaque formation, plaque maturation, and plaque destabilization and eventual rupture [30].
Patients with RA, as well as those with SLE, have increased mortality compared to the general population [31,32]. Increased mortality from CVD has been reported in epidemiologic studies that have focused on RA [33]. In similarity to RA, the risk of CVD-related death is increased in SLE [31,34].
In prospective studies, the incidence rates of myocardial infarction and stroke in patients with SLE have been found to be high. The relative risk of myocardial infarction or stroke compared to the normal population is approximately 2–3 [35,36,37]. The highest relative risks have been reported for premenopausal women (8–50-fold higher risk), early in the course of SLE (<1 year after diagnosis, risk increased 4–10-fold), and in patients with renal involvement (4–18-fold higher risk) [38,39,40]. Other studies have focused on examining the incidence of CVD in patients with SLE compared to the expected CVD incidence, based on the presence of traditional risk factors. Even here, the incidence of CVD has been found to be considerably higher than expected [41,42]. In addition, risk of mortality post-myocardial infarction seems to be higher in patients with SLE than in the normal population, at least in the short term, while the long-term risk of mortality post-stroke is also increased [43,44].
Approximately 30% of patients with SLE display antiphospholipid antibodies (at least one of the following: anticardiolipin or anti-β2-glycoprotein-I antibodies, or a positive lupus anticoagulant test) and about 15–20% suffer from antiphospholipid syndrome (APS), which is characterized by an increased risk of thromboembolic disease and/or pregnancy morbidity. Ischemic stroke is the most common arterial manifestation of APS, while myocardial infarction is less common [45].
Whereas some studies have focused on CRP levels as a risk factor for future cardiovascular events in RA, studies of CRP levels in patients with SLE in relation to risks of CVD or stroke are scarce [46,47,48,49]. Statin therapy is likely to be safe and seems to result in significant reduction of plasma CRP concentrations in patients with SLE [50]. For patients with SLE, the Systemic Lupus International Collaborating Clinics/American College of Rheumatology (SLICC/ACR) Damage Index (SDI) constitutes a validated instrument to assess irreversible organ damage, including myocardial infarction and stroke [51]. We identified two studies in which CRP levels were analyzed in relation to accrual of damage. In the Hopkins Lupus cohort, Lee et al. showed that serum CRP levels (measured with the high-sensitivity technique) were independently associated with the total SDI score, although not specifically for myocardial infarction or stroke [52]. Our group has reported a similar association between CRP and global SDI [53]. Furthermore, in the SLICC cohort, we evaluated whether CRP could be predictive in terms of future damage accrual but obtained negative results [54].
4. Immunoregulatory Functions of CRP and other Pentraxins in SLE
The high accumulation of apoptotic cell debris and the formation of antinuclear antibodies (ANA), together with dysfunctional elimination of immune complexes are all key features of SLE pathogenesis [55]. In this context, it is of particular interest that CRP immune function can be viewed as a less specific albeit rapidly produced innate ancestor version of the phylogenetically more recent antibodies of adaptive immunity [56]. CRP is a pattern-recognition molecule of the innate immune system, and its binding to ligands such as surface-exposed phosphorylcholine on, for example, cellular debris can mediate direct prophagocytotic opsonization [57] and interactions with immunoglobulin receptors (Fc receptors) [56], as well as trigger ‘classical’ complement activation [58]. The latter promotes additional opsonization through subsequent covalent surface-binding of activated complement proteins.
Immune complex clearance is generally supported by efficient classical complement activation, and SLE pathogenesis is indeed intimately related to this activation pathway. Although homozygous complement deficiencies are extremely rare, they tell us a great deal about the normal physiological activities of the complement system in humans [59]. Homozygous genetic deficiencies in the initial proteins of the classical complement pathway (C1 proteins) are linked to a very high risk of developing SLE [60], and single nucleotide polymorphisms of the CRP gene are associated with ANA formation and SLE, possibly via the lowering of CRP levels [12].
In vertebrates, surface binding via the recognition face of the CRP molecule activates the calcium-dependent classical arm of the complement cascade by binding complement protein 1q (C1q) via its effector face [61,62]. C1q binding to the mCRP isoform has been demonstrated [63], and mCRP is capable of supporting complement-dependent phagocytosis and the oxidative burst in phagocytes [64]. Unlike immunoglobulin G-triggered classical activation, CRP-mediated initiation of the classical route typically does not proceed to the membrane-attack complex-forming ‘terminal’ stage of complement activation [65]. This is most likely due to direct interactions of CRP with inhibitory complement regulators. It is well-established that CRP can bind to the soluble complement inhibitor factor H without compromising its inhibitory function, thereby limiting the continued activation of complement via the convertases, by accelerating their decay [66,67,68,69] and by serving as co-factor for Factor I in cleaving surface-bound C3b [70,71]. In addition, surface-bound mCRP can bind Factor H and, thereby, modulate complement activation [72]. Anti-C1q autoantibodies are frequently detected in lupus nephritis (LN) [73,74] and it is possible that autoantibodies targeting other proteins linked to classical complement activation, e.g., CRP, could affect the complement-mediated clearance of cellular debris [75,76]. CRP (and/or PTX3), complement, and immunoglobulins may co-localize with electron-dense deposits in glomerular LN [77,78]. Furthermore, it is possible that pre-immunization with pentraxins, leading to the triggering of anti-PTX3 antibody development, prevents progression to LN [77]. Anti-CRP antibodies appear to target mainly the motifs of mCRP (further described in Section 7) and are typically associated with LN. The mCRP amino acids 35–47 have been reported to represent an autoantibody target motif that is especially prone to anti-CRP binding in LN. From the complement-immunomodulatory point-of-view, it is interesting to note that this epitope also facilitates factor H binding and activity—which could be reversed by anti-CRP antibodies [79]. In accordance with this, factor H levels are low in LN and factor H dysregulation and polymorphisms are associated with active nephritis [80,81]. Other members of the factor H family, i.e., factor H-related Proteins 1 and 5, have recently been shown to be capable of binding DNA and subsequently recruiting mCRP and enhancing complement activation [82]. In addition, factor H-related Protein 4 has been reported to bind pCRP [83,84].
Ligand-bound CRP on necrotic cells and/or otherwise immobilized CRP can recruit the classical pathway inhibitor C4-binding protein (C4bp) while retaining the complement-inhibitory activity of C4bp [85,86]. It is possible that this C4bp–CRP interaction limits CRP–C1q binding [85,86], and thereby subsequent classical activation. Classical complement activation triggered by ligand-bound CRP may be downregulated during substantial increases in the concentrations of CRP, presumably through humoral CRP–C1q consumption [63,87]. This could also be of pathophysiologic relevance in SLE, where the CRP levels can be low despite active inflammation. Related to the now well-established role of collectins in the ‘lectin’ activation pathway of complement, a potential role of CRP in conveying C1q-dependent complement activation by collectin Placenta 1 has been reported [88]. This is a topic that would be interesting to pursue further in relation to autoimmune diseases.
In similarity to IgG, surface-bound CRP (and other pentraxins) can bind directly to all Fcγ-receptors [89], potentially activating phagocytes and facilitating elimination via phagocytosis, which is highly relevant for waste disposal mechanisms. The low-affinity FcγRIIa (CD32) has emerged as the primary functional CRP receptor [56,89,90,91]. However, unlike IgG, the CRP–FcγRIIa interaction depends on the R allele of the receptor polymorphism at amino acid 131 [92]. Since SLE pathogenesis is linked to immune complex-induced production of IFN-α by plasmacytoid dendritic cells, it is highly interesting to note that FcγRIIa also mediates the initial internalization of immune complexes that prompts intracellular TLR activation and activation of IFN-α [93]. Considering the protective effects of CRP seen in animal models of lupus, it is tempting to speculate that CRP acts as a modulator of IFN-α production by altering the immune complex handling by plasma-cytoid dendritic cells. Accordingly, Mold and Du Clos reported that CRP indeed inhibits such immune complex-triggered activation of IFN-α, although the mechanism appeared to involve instead the endosomal processing of immune complexes [94]. Additional mechanistic studies on the CRP-mediated downregulation of immune complex-triggered IFN-α in SLE are highly warranted. Another intriguing finding that merits further attention is the potentially immunomodulatory effect of the CRP interaction with FcαRI, the IgA receptor [95].
5. Regulation of CRP Synthesis in SLE
Hepatocytes are considered the major source of CRP, although extrahepatic syntheses have been reported [96,97,98,99,100]. The CRP gene is located on chromosome 1q23.2 and hepatic production of CRP is mainly regulated at the transcriptional level, with IL-6 and IL-1β being the most important inducers [101,102]. IL-6 signaling in hepatocytes mediates the activation and CRP promoter-binding of signal inducer and activator of transcription 3 (STAT3) [102] and the CCAAT/enhancer binding protein β (C/EBPβ) [101,103]. In hepatic cell lines, the addition of IL-1β and subsequent NF-κB activation are usually required for CRP transcription, whereas in primary hepatocytes, IL-6 is sufficient for CRP production [101,104,105].
Although CRP is generally an excellent biomarker of inflammation and tissue damage due to its massive increase in level upon IL-6 induction, it is not useful in all inflammatory conditions. SLE represents an exception, in that the CRP levels rarely mirror the disease activity [15,106]. Inflammatory myopathies, primary Sjögren’s syndrome and systemic sclerosis are other diseases for which CRP is considered an unreliable marker for monitoring disease activity [107]. In addition, viral infections rarely exhibit a substantial rise in CRP levels [108].
The above-mentioned conditions all have in common the activation of Type I IFNs. The most widely studied Type I IFN is IFN-α, which comprises 12 subtypes. Apart from having a physiologic function in defense against viruses, IFN-α induces and maintains autoimmune pathology through facilitation of autoantibody production and many other functions, as reviewed elsewhere [109]. Receptors for Type I IFNs (IFN-α /β receptor; IFNAR) are ubiquitously expressed and mediate the activation of different STAT heterodimers and homodimers for the activation of antiviral, inflammatory and regulatory gene expression [110]. Already in 2008, Type I IFNs were highlighted as potential inhibitors of CRP production via their activation of STAT1, so as to counteract the STAT3 effects, and/or the activation of an inhibitory isoform of C/EBPβ [10]. Later, an inhibitory effect of IFN-α (all subtypes) on CRP transcription and production was indeed shown in a hepatic cell line and in primary hepatocytes, respectively [104]. Further in vivo studies of CRP levels and IFN-α levels in patients with SLE have lent support to the notion of a regulatory role for IFN-α in CRP production [54,111], although the exact intracellular pathways remain unknown.
Polymorphisms of the CRP gene have been linked to differences in basal CRP levels and the risk of SLE and/or cardiovascular events [12,112,113,114]. One of these polymorphisms, rs1205, has been studied together with IL-6 and IFN-α with respect to the impact of these potential regulators of CRP levels in SLE, revealing lower CRP levels in patients with IFNα activation and/or the CRP-lowering polymorphism rs1205 (Figure 1). Thus, the relative lack of CRP response seen in viral infections and Type I IFN-driven autoimmune diseases can be attributed to an IFN-α-dependent downregulation of CRP transcription, as well as CRP gene polymorphisms, which are over-represented among patients with SLE [111,115].
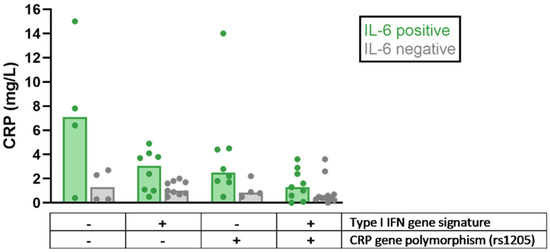
Figure 1.
Differences in C-reactive protein (CRP) levels among patients with systemic lupus erythematosus (SLE) stratified based on the presence of detectable interleukin 6 (IL-6) levels, Type I interferon (IFN) gene signature, and CRP-lowering gene polymorphism (rs1205), respectively. All patients had a low disease activity but could be serologically active at sampling. Bars indicate median values. Dots represent individual values. Data shown in the figure were adopted from Enocsson et al. [115], with permission from Frontiers Media, 2021 (Creative Commons Attribution licence, version 4.0).
6. Structural Isoforms of CRP with Distinctive Biologic Effects
As mentioned above, pCRP can under certain conditions dissociate irreversibly into the monomeric form (mCRP), which displays distinctly different conformational characteristics and antigenic epitopes [116,117]. Emerging data implicate mCRP as the main CRP isoform that regulates local inflammatory processes [118,119,120]. Furthermore, mCRP may bind to IgG-containing immune complexes and facilitate silent Fc receptor-mediated removal via the reticuloendothelial system and complement deficiencies may result immune complex deposition outside the reticuloendothelial system [121,122].
Cell death occurs during inflammation, and the damaged cell membrane in apoptosis or necrosis is the main target of CRP recognition [4]. Using electron microscopy, the detection of new epitopes of the antigen, and immunofluorescence colocalization, Ji et al. have shown that the binding of CRP to the damaged cell membrane induces rapid transformation to mCRP, and that this dissociation process is accompanied by significant enhancement of complement activation and cellular stimulation capacity [118]. Eisenhardt et al. obtained similar results with activated platelet membranes [123]. In addition, inflammatory conditions such as moderate acidification and oxidative stress also promote conformational switching from pCRP to mCRP.
During acute inflammatory cardiovascular events, such as thrombosis and myocardial infarction, the CRP levels increase rapidly, while activated platelets in blood vessels or cell necrosis caused by hypoxia in the heart provide abundant damaged membrane ligands for CRP dissociation, which leads to the accumulation of a large amount of ’active’ mCRP in the lesions within a short period of time [118,124]. This results in the excessive activation of neutrophils, platelets, monocytes and complement, thereby exacerbating the inflammation [118,123,125,126,127,128,129,130]. Furthermore, the conversion of pCRP to mCRP has been observed on microparticles in the blood obtained from patients who suffered myocardial infarctions, as well as on beta-amyloid plaques [131,132]. This process indicates a physiologic mechanism of CRP isomerization that is driven by the inflammatory microenvironment and, at the same time, supports the concept of mCRP occurring as a natural isomer of CRP and being involved in regulating inflammatory processes [120,133].
Most of the abovementioned dissociation scenarios for generating mCRP are specifically linked to inflammation. Thus, it is plausible that mCRP is generated predominately within the inflamed local tissue. Based on the strong proinflammatory activities of mCRP, we propose that, in addition to being an activating mechanism, the conversion of pCRP to mCRP serves as a buffering mechanism that localizes the proinflammatory actions to the site of the inflammation [118]. This mechanism could protect the body from systemic challenge in response to increased circulating levels of pCRP. It is worth noting that the bioactivities of mCRP largely overlap with, and occasionally exceed, those previously ascribed to pCRP. These bioactivities of mCRP include the activation of complement and the stimulation of endothelial cells, neutrophils and platelets, as well as its binding to ligands, e.g., LDL, C1q and factor H [63,72,85,126,128,134,135,136,137,138,139,140,141,142,143]. This raises the possibility that some of the reported actions of pCRP originate from mCRP formed during the purification process and/or storage.
The allosteric switch from pCRP, as a marker of inflammation, to functional mCRP that actually participates in the inflammatory process, enables this acute-phase protein to play active roles in a controlled manner under different pathophysiologic conditions. Thus, CRP can be regarded as a potential fine tuner of inflammation. Although there have been long-term debates about the biological significance of mCRP, recent studies have revealed the pathway of mCRP production, the regulatory effects of mCRP on innate and adaptive humoral immunity and inflammatory processes, and the presence of mCRP in focal tissues [63,118,123,126,128,134,135,136,137,138,142,144,145].
Interfering with the dissociation of CRP and the way in which mCRP exerts its biologic functions are candidate pathways towards designing treatment strategies for CVD. Since the specific contribution of mCRP depends on the inflammatory microenvironment, a clear understanding of the molecular mechanisms that act in different pathophysiologic conditions is a prerequisite for the design and selection of appropriate interventions. Several important issues remain to be resolved: (1) how to establish either direct or indirect detection methods that use mCRP as a disease marker; (2) how to establish an association between mCRP and disease processes; (3) how to describe the short-and long-term response profiles of different cell types to mCRP in a systematic way; and (4) identification of the receptor (s) that mediate the downstream effects of mCRP in lipid rafts.
7. Autoantibodies Directed against CRP in SLE and Related Conditions
Already in the mid-1980s, the presence of autoantibodies against CRP was described and linked to the debilitated ability of CRP to solubilize chromatin in a patient with SLE [146]. Subsequently, Bell et al. reported a high frequency of IgG antibodies to cryptic epitopes of CRP, first in patients suffering from the ‘autoimmune-like’ toxic oil syndrome and thereafter in patients with SLE [147,148]. Similarly, we have shown a prevalence of anti-CRP antibodies of approximately 40% in patients with SLE, with a distinct positive correlation between antibody occurrence/concentration and disease activity.
In our first study, we demonstrated that some patients with SLE were anti-CRP antibody positive on one occasion but negative on another occasion [149]. In succeeding investigations, we analyzed the antibody levels in consecutive samples from 10 well-charac-terized patients with SLE and showed that the levels of anti-CRP antibodies paralleled the clinical disease activity, usually with high levels of these antibodies appearing during disease flares [150]. In total, 70% of the patients were positive for anti-CRP antibodies on at least one occasion, and the levels correlated with disease activity assessed using the SLE disease activity index (SLEDAI).
Our findings were essentially confirmed by Rosenau and Schur, who demonstrated the presence of antibodies against CRP in the sera obtained from patients with different rheumatologic conditions, including SLE, where they observed an autoantibody frequency of 23% [151]. However, in our hands, sera from patients with RA or inflammatory bowel disease have consistently been negative in the anti-CRP assay, whereas a few additional patients with primary Sjögren’s syndrome and chronic hepatitis C infection tested positive [149,152]. Others have found anti-CRP antibodies in patients with tubulointerstitial nephritis and uveitis (TINU) syndrome [153]. Furthermore, Figueredo et al. have demonstrated the presence of anti-CRP antibodies in patients with SLE with or without APS; the anti-CRP-positive cases with SLE had lower C3 levels and were more likely to have anti-dsDNA and anticardiolipin antibodies as compared to the anti-CRP antibody-negative individuals. In addition, the frequency of LN was higher among the anti-CRP antibody positive SLE cases [154]. The biological properties of anti-CRP antibodies have also been investigated. Janko et al. demonstrated that anti-CRP—as well as anti-dsDNA-antibodies bind to apoptotic materials and, via clearance by macrophages, induce a pro-inflammatory cytokine response [75].
More recently, a large longitudinal study from Europe identified the presence of anti-CRP antibodies at the onset of LN as a strong risk factor for a composite outcome of non-response, renal flare, and end-stage renal disease after 2 years of standard LN treatment [155]. Analyses of the antigen specificity of the anti-CRP assay have revealed that autoantibodies to CRP in SLE are directed towards hidden epitopes, or neo-epitopes, of CRP (e.g., mCRP), and that immune complexes isolated from SLE sera do not induce false positive anti-CRP antibody test results [79,156,157]. Thus, in similarity to anti-C1q antibodies in SLE, reacting exclusively with an epitope that is exposed on structurally modified C1q [59,158], anti-CRP antibodies bind to mCRP on cells, as well as on tissues and in solution [76].
8. Conclusions
Even though almost a century has passed since the discovery of CRP, the biological effects of this highly conserved molecule are still poorly understood. Nonetheless, emerging data highlight the importance of structural isoforms of CRP and their associations with the complement system and CVD. As summarized in Figure 2, CRP plays a complex role in SLE—a disease in which CRP, in contrast to most other rheumatic conditions, constitutes an unreliable biomarker of inflammation. Recent data indicate that the combined effects of genetics and the Type I IFN signature are responsible for the dissociated correlation between CRP and IL-6 levels in patients with SLE. Given the potential activities of CRP in facilitating the removal of apoptotic debris and immune complexes, this may be of high relevance in terms of driving LN and the accrual of organ damage in SLE.
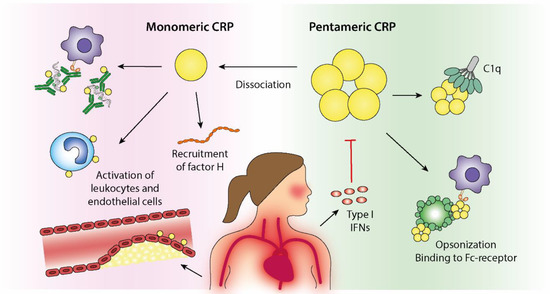
Figure 2.
Immunoregulatory effects of pentameric C-reactive protein (pCRP) and monomeric CRP (mCRP) in the context of systemic lupus erythematosus (SLE) and cardiovascular disease. Dissociation of pCRP to mCRP will take place in inflammatory conditions and at cell surfaces and results in local immunoregulatory effects. The biological properties of mCRP partly overlaps the pCRP effects but is generally ascribed a more active and proinflammatory profile. CRP binds to and opsonizes dying cells and cell remnants, which facilitates phagocytosis via Fc-receptor binding. Furthermore, CRP activates classical complement activation via its binding to C1q, resulting in increased opsonization by C3b. Recruitment of Factor H will however limit progression of the complement cascade to membrane attack complex formation. Increased levels of CRP can therefore contribute to efficient clearance of potential autoantigens and thus, be beneficial in autoimmune conditions. The ability of CRP to facilitate immune complex elimination further implies a protective role of CRP in autoimmune diseases. However, increased Type I IFN activity, frequently observed in patients with SLE, inhibits CRP production, which theoretically could increase the autoantigen burden and disease activity. Proatherogenic and protrombotic effects of CRP are attributed to its stimulation of endothelial cells, neutrophils and platelets.
Author Contributions
Conceptualization, C.S.; search strategy and review of publications, H.E., J.K., H.-Y.L., Y.W., J.W. and C.S.; draft preparation, review and editing, H.E., J.K., H.-Y.L., Y.W., I.K., J.W. and C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from Region Östergötland (ALF Grants), the Swedish Rheumatism Association, the Ulla and Roland Gustafsson Foundation, the King Gustaf V’s 80-year Anniversary foundation and the King Gustaf V and Queen Victoria’s Freemasons foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors thank Lina Wirestam for the schematic illustration (Figure 2).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tillett, W.S.; Francis, T. Serological Reactions in Pneumonia with a Non-Protein Somatic Fraction of Pneumococcus. J. Exp. Med. 1930, 52, 561–571. [Google Scholar] [CrossRef]
- Kushner, I.; Samols, D. Oswald Avery and the pneumococcus. Pharos Alpha Omega Alpha Honor Med. Soc. 2011, 74, 14–18. [Google Scholar]
- Volanakis, J.E.; Kaplan, M.H. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc. Soc. Exp. Biol. Med. 1971, 136, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Invest. 2003, 111, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, X.; Zou, H.; Dai, Z.; Feng, S.; Zhang, M.; Xiao, G.; Liu, Z.; Cheng, Q. The Basic Characteristics of the Pentraxin Family and Their Functions in Tumor Progression. Front Immunol. 2020, 11, 1757. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Potempa, L.A.; El Kebir, D.; Filep, J.G. C-reactive protein and inflammation: Conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Brilland, B.; Vinatier, E.; Subra, J.F.; Jeannin, P.; Augusto, J.F.; Delneste, Y. Anti-Pentraxin Antibodies in Autoimmune Diseases: Bystanders or Pathophysiological Actors? Front Immunol. 2021, 11, 626343. [Google Scholar] [CrossRef]
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar] [CrossRef]
- Gabay, C.; Roux-Lombard, P.; de Moerloose, P.; Dayer, J.M.; Vischer, T.; Guerne, P.A. Absence of correlation between interleukin 6 and C-reactive protein blood levels in systemic lupus erythematosus compared with rheumatoid arthritis. J. Rheumatol. 1993, 20, 815–821. [Google Scholar]
- Gaitonde, S.; Samols, D.; Kushner, I. C-reactive protein and systemic lupus erythematosus. Arthritis. Rheum. 2008, 59, 1814–1820. [Google Scholar] [CrossRef]
- Kuhlenbaeumer, G.; Huge, A.; Berger, K.; Kessler, C.; Voelzke, H.; Funke, H.; Stoegbauer, F.; Stoll, M.; Ringelstein, E.B. Genetic variants in the C-reactive protein gene are associated with microangiopathic ischemic stroke. Cerebrovasc Dis. 2010, 30, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.I.; Cunninghame Graham, D.S.; Shepherd, C.; Roberton, C.A.; Whittaker, J.; Meeks, J.; Powell, R.J.; Isenberg, D.A.; Walport, M.J.; Vyse, T.J. Polymorphism at the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Hum. Mol. Genet. 2004, 13, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, M.; Kushner, I. It’s time to redefine inflammation. FASEB J. 2017, 31, 1787–1791. [Google Scholar] [CrossRef]
- Lindahl, B.; Toss, H.; Siegbahn, A.; Venge, P.; Wallentin, L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. N. Engl. J. Med. 2000, 343, 1139–1147. [Google Scholar] [CrossRef]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Wirestam, L.; Pihl, S.; Saleh, M.; Wetterö, J.; Sjöwall, C. Plasma C-Reactive Protein and Pentraxin-3 Reference Intervals During Normal Pregnancy. Front. Immunol. 2021, 12, 722118. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.; Gladman, D.; Helliwell, P.; Marchesoni, A.; Mease, P.; Mielants, H.; Group, C.S. Classification criteria for psoriatic arthritis: Development of new criteria from a large international study. Arthritis. Rheum. 2006, 54, 2665–2673. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, B.; Agueda, A.; Monti, S.; Buttgereit, F.; de Boysson, H.; Brouwer, E.; Cassie, R.; Cid, M.C.; Dasgupta, B.; Dejaco, C.; et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann. Rheum. Dis. 2020, 79, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Cimmino, M.A.; Kremers, H.M.; Schmidt, W.A.; Schirmer, M.; Salvarani, C.; Bachta, A.; Dejaco, C.; Duftner, C.; Jensen, H.S.; et al. 2012 Provisional classification criteria for polymyalgia rheumatica: A European League Against Rheumatism/American College of Rheumatology collaborative initiative. Arthritis. Rheum. 2012, 64, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Kronbichler, A.; Shin, J.I.; Lee, K.H.; Nakagomi, D.; Quintana, L.F.; Busch, M.; Craven, A.; Luqmani, R.A.; Merkel, P.A.; Mayer, G.; et al. Clinical associations of renal involvement in ANCA-associated vasculitis. Autoimmun. Rev. 2020, 19, 102495. [Google Scholar] [CrossRef] [PubMed]
- Morrow, W.J.; Isenberg, D.A.; Parry, H.F.; Snaith, M.L. C-reactive protein in sera from patients with systemic lupus erythematosus. J. Rheumatol. 1981, 8, 599–604. [Google Scholar] [PubMed]
- Bianchi, M.; Kozyrev, S.V.; Notarnicola, A.; Hultin Rosenberg, L.; Karlsson, A.; Pucholt, P.; Rothwell, S.; Alexsson, A.; Sandling, J.K.; Andersson, H.; et al. Contribution of rare genetic variation to disease susceptibility in a large Scandinavian myositis cohort. Arthritis. Rheumatol. 2021. Epub Ahead of Print. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Almlof, J.C.; Leonard, D.; Sjöwall, C.; Syvanen, A.C.; Rönnblom, L.; Sandling, J.K.; Nordmark, G. Shared and Unique Patterns of DNA Methylation in Systemic Lupus Erythematosus and Primary Sjogren’s Syndrome. Front Immunol. 2019, 10, 1686. [Google Scholar] [CrossRef] [PubMed]
- Strangfeld, A.; Eveslage, M.; Schneider, M.; Bergerhausen, H.J.; Klopsch, T.; Zink, A.; Listing, J. Treatment benefit or survival of the fittest: What drives the time-dependent decrease in serious infection rates under TNF inhibition and what does this imply for the individual patient? Ann. Rheum. Dis. 2011, 70, 1914–1920. [Google Scholar] [CrossRef]
- Askling, J. Malignancy and rheumatoid arthritis. Curr. Rheumatol. Rep. 2007, 9, 421–426. [Google Scholar] [CrossRef]
- Manger, B.; Schett, G. Paraneoplastic syndromes in rheumatology. Nat. Rev. Rheumatol. 2014, 10, 662–670. [Google Scholar] [CrossRef]
- Ogata, A.; Kato, Y.; Higa, S.; Yoshizaki, K. IL-6 inhibitor for the treatment of rheumatoid arthritis: A comprehensive review. Mod. Rheumatol. 2019, 29, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef]
- de Ferranti, S.D.; Rifai, N. C-reactive protein: A nontraditional serum marker of cardiovascular risk. Cardiovasc. Pathol. 2007, 16, 14–21. [Google Scholar] [CrossRef]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis. Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.A.; Chang, S.C.; Liao, K.P.; Lu, B.; Fine, A.R.; Solomon, D.H.; Costenbader, K.H.; Karlson, E.W. Rheumatoid Arthritis and Mortality Among Women During 36 Years of Prospective Follow-Up: Results From the Nurses’ Health Study. Arthritis. Care Res. 2016, 68, 753–762. [Google Scholar] [CrossRef]
- Avina-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis. Rheum. 2008, 59, 1690–1697. [Google Scholar] [CrossRef]
- Björnadal, L.; Yin, L.; Granath, F.; Klareskog, L.; Ekbom, A. Cardiovascular disease a hazard despite improved prognosis in patients with systemic lupus erythematosus: Results from a Swedish population based study 1964-95. J. Rheumatol. 2004, 31, 713–719. [Google Scholar] [PubMed]
- Arkema, E.V.; Svenungsson, E.; Von Euler, M.; Sjöwall, C.; Simard, J.F. Stroke in systemic lupus erythematosus: A Swedish population-based cohort study. Ann. Rheum. Dis. 2017, 76, 1544–1549. [Google Scholar] [CrossRef]
- Avina-Zubieta, J.A.; To, F.; Vostretsova, K.; De Vera, M.; Sayre, E.C.; Esdaile, J.M. Risk of Myocardial Infarction and Stroke in Newly Diagnosed Systemic Lupus Erythematosus: A General Population-Based Study. Arthritis. Care Res. 2017, 69, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, I.; Parodis, I.; Gunnarsson, I.; Zickert, A.; Hofman-Bang, C.; Wallen, H.; Svenungsson, E. Myocardial infarctions, subtypes and coronary atherosclerosis in SLE: A case-control study. Lupus. Sci. Med. 2021, 8, e000515. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, C.; Ohman, M.L.; Nived, O.; Rantapaa Dahlqvist, S. Cardiovascular event in systemic lupus erythematosus in northern Sweden: Ancidence and predictors in a 7-year follow-up study. Lupus 2012, 21, 452–459. [Google Scholar] [CrossRef]
- Hermansen, M.L.; Lindhardsen, J.; Torp-Pedersen, C.; Faurschou, M.; Jacobsen, S. The risk of cardiovascular morbidity and cardiovascular mortality in systemic lupus erythematosus and lupus nephritis: A Danish nationwide population-based cohort study. Rheumatology 2017, 56, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A., Jr.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: Comparison with the Framingham Study. Am. J. Epidemiol. 1997, 145, 408–415. [Google Scholar] [CrossRef]
- Esdaile, J.M.; Abrahamowicz, M.; Grodzicky, T.; Li, Y.; Panaritis, C.; du Berger, R.; Cote, R.; Grover, S.A.; Fortin, P.R.; Clarke, A.E.; et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis. Rheum. 2001, 44, 2331–2337. [Google Scholar] [CrossRef]
- Fischer, L.M.; Schlienger, R.G.; Matter, C.; Jick, H.; Meier, C.R. Effect of rheumatoid arthritis or systemic lupus erythematosus on the risk of first-time acute myocardial infarction. Am. J. Cardiol. 2004, 93, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.R.; Liu, C.W.; Wu, Y.W.; Lai, K.R.; Wu, C.Y.; Lin, J.W.; Chan, C.L.; Pan, R.H. Systemic lupus erythematosus is associated with poor outcome after acute myocardial infarction. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 1400–1407. [Google Scholar] [CrossRef]
- Rossides, M.; Simard, J.F.; Svenungsson, E.; von Euler, M.; Arkema, E.V. Mortality and Functionality after Stroke in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2017, 44, 1590–1596. [Google Scholar] [CrossRef]
- Svenungsson, E.; Antovic, A. The antiphospholipid syndrome - often overlooked cause of vascular occlusions? J. Intern. Med. 2020, 287, 349–372. [Google Scholar] [CrossRef]
- Goodson, N.J.; Symmons, D.P.; Scott, D.G.; Bunn, D.; Lunt, M.; Silman, A.J. Baseline levels of C-reactive protein and prediction of death from cardiovascular disease in patients with inflammatory polyarthritis: A ten-year followup study of a primary care-based inception cohort. Arthritis. Rheum. 2005, 52, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Innala, L.; Möller, B.; Ljung, L.; Magnusson, S.; Smedby, T.; Södergren, A.; Ohman, M.L.; Rantapaa-Dahlqvist, S.; Wallberg-Jonsson, S. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: A five year prospective study. Arthritis Res. Ther. 2011, 13, R131. [Google Scholar] [CrossRef] [PubMed]
- Maradit-Kremers, H.; Nicola, P.J.; Crowson, C.S.; Ballman, K.V.; Gabriel, S.E. Cardiovascular death in rheumatoid arthritis: A population-based study. Arthritis. Rheum. 2005, 52, 722–732. [Google Scholar] [CrossRef]
- Kao, A.H.; Wasko, M.C.; Krishnaswami, S.; Wagner, J.; Edmundowicz, D.; Shaw, P.; Cunningham, A.L.; Danchenko, N.; Sutton-Tyrrell, K.; Tracy, R.P.; et al. C-reactive protein and coronary artery calcium in asymptomatic women with systemic lupus erythematosus or rheumatoid arthritis. Am. J. Cardiol. 2008, 102, 755–760. [Google Scholar] [CrossRef]
- Sahebkar, A.; Rathouska, J.; Derosa, G.; Maffioli, P.; Nachtigal, P. Statin impact on disease activity and C-reactive protein concentrations in systemic lupus erythematosus patients: A systematic review and meta-analysis of controlled trials. Autoimmun. Rev. 2016, 15, 344–353. [Google Scholar] [CrossRef]
- Gladman, D.; Ginzler, E.; Goldsmith, C.; Fortin, P.; Liang, M.; Urowitz, M.; Bacon, P.; Bombardieri, S.; Hanly, J.; Hay, E.; et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis. Rheum. 1996, 39, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Singh, S.; Link, K.; Petri, M. High-sensitivity C-reactive protein as an associate of clinical subsets and organ damage in systemic lupus erythematosus. Semin. Arthritis. Rheum. 2008, 38, 41–54. [Google Scholar] [CrossRef]
- Enocsson, H.; Wetterö, J.; Skogh, T.; Sjöwall, C. Soluble urokinase plasminogen activator receptor levels reflect organ damage in systemic lupus erythematosus. Transl. Res. 2013, 162, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Enocsson, H.; Wirestam, L.; Dahle, C.; Padyukov, L.; Jonsen, A.; Urowitz, M.B.; Gladman, D.D.; Romero-Diaz, J.; Bae, S.C.; Fortin, P.R.; et al. Soluble urokinase plasminogen activator receptor (suPAR) levels predict damage accrual in patients with recent-onset systemic lupus erythematosus. J. Autoimmun. 2020, 106, 102340. [Google Scholar] [CrossRef]
- Dieker, J.; Berden, J.H.; Bakker, M.; Briand, J.P.; Muller, S.; Voll, R.; Sjöwall, C.; Herrmann, M.; Hilbrands, L.B.; van der Vlag, J. Autoantibodies against Modified Histone Peptides in SLE Patients Are Associated with Disease Activity and Lupus Nephritis. PLoS ONE 2016, 11, e0165373. [Google Scholar] [CrossRef]
- Lu, J.; Marnell, L.L.; Marjon, K.D.; Mold, C.; Du Clos, T.W.; Sun, P.D. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 2008, 456, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Ganrot, P.O.; Kindmark, C.O. C-reactive protein--a phagocytosis-promoting factor. Scand J. Clin. Lab. Investig. 1969, 24, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, R.F.; Osmand, A.P.; Lint, T.F.; Gewurz, H. Interaction of C-reactive protein with lymphocytes and monocytes: Complement-dependent adherence and phagocytosis. J. Immunol. 1976, 117, 774–781. [Google Scholar]
- Manderson, A.P.; Botto, M.; Walport, M.J. The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 2004, 22, 431–456. [Google Scholar] [CrossRef]
- Truedsson, L.; Bengtsson, A.A.; Sturfelt, G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity 2007, 40, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.H.; Volanakis, J.E. Interaction of C-reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal C-polysaccharide and with the choline phosphatides, lecithin and sphingomyelin. J. Immunol. 1974, 112, 2135–2147. [Google Scholar]
- Siegel, J.; Rent, R.; Gewurz, H. Interactions of C-reactive protein with the complement system. I. Protamine-induced consumption of complement in acute phase sera. J. Exp. Med. 1974, 140, 631–647. [Google Scholar] [CrossRef]
- Ji, S.R.; Wu, Y.; Potempa, L.A.; Liang, Y.H.; Zhao, J. Effect of modified C-reactive protein on complement activation: A possible complement regulatory role of modified or monomeric C-reactive protein in atherosclerotic lesions. Arterioscler Thromb. Vasc. Biol. 2006, 26, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Zeller, J.; Bogner, B.; Kiefer, J.; Braig, D.; Winninger, O.; Fricke, M.; Karasu, E.; Peter, K.; Huber-Lang, M.; Eisenhardt, S.U. CRP Enhances the Innate Killing Mechanisms Phagocytosis and ROS Formation in a Conformation and Complement-Dependent Manner. Front. Immunol. 2021, 12, 721887. [Google Scholar] [CrossRef]
- Berman, S.; Gewurz, H.; Mold, C. Binding of C-reactive protein to nucleated cells leads to complement activation without cytolysis. J. Immunol. 1986, 136, 1354–1359. [Google Scholar]
- Jarva, H.; Jokiranta, T.S.; Hellwage, J.; Zipfel, P.F.; Meri, S. Regulation of complement activation by C-reactive protein: Targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8–11. J. Immunol. 1999, 163, 3957–3962. [Google Scholar]
- Mold, C.; Gewurz, H.; Du Clos, T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology 1999, 42, 23–30. [Google Scholar] [CrossRef]
- Weiler, J.M.; Daha, M.R.; Austen, K.F.; Fearon, D.T. Control of the amplification convertase of complement by the plasma protein beta1H. Proc. Natl. Acad. Sci USA 1976, 73, 3268–3272. [Google Scholar] [CrossRef] [PubMed]
- Whaley, K.; Ruddy, S. Modulation of the alternative complement pathways by beta 1 H globulin. J. Exp. Med. 1976, 144, 1147–1163. [Google Scholar] [CrossRef]
- Harrison, R.A.; Lachmann, P.J. The physiological breakdown of the third component of human complement. Mol. Immunol. 1980, 17, 9–20. [Google Scholar] [CrossRef]
- Pangburn, M.K.; Schreiber, R.D.; Muller-Eberhard, H.J. Human complement C3b inactivator: Isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J. Exp. Med. 1977, 146, 257–270. [Google Scholar] [CrossRef]
- Mihlan, M.; Stippa, S.; Jozsi, M.; Zipfel, P.F. Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ. 2009, 16, 1630–1640. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, X.W.; Song, Y.; Yu, F.; Zhao, M.H. Anti-C1q autoantibodies from active lupus nephritis patients could inhibit the clearance of apoptotic cells and complement classical pathway activation mediated by C1q in vitro. Immunobiology 2014, 219, 980–989. [Google Scholar] [CrossRef]
- Trendelenburg, M.; Marfurt, J.; Gerber, I.; Tyndall, A.; Schifferli, J.A. Lack of occurrence of severe lupus nephritis among anti-C1q autoantibody-negative patients. Arthritis. Rheum. 1999, 42, 187–188. [Google Scholar] [CrossRef]
- Janko, C.; Franz, S.; Munoz, L.E.; Siebig, S.; Winkler, S.; Schett, G.; Lauber, K.; Sheriff, A.; van der Vlag, J.; Herrmann, M. CRP/anti-CRP antibodies assembly on the surfaces of cell remnants switches their phagocytic clearance toward inflammation. Front. Immunol. 2011, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Sjöwall, C.; Wetterö, J. Pathogenic implications for autoantibodies against C-reactive protein and other acute phase proteins. Clin. Chim. Acta. 2007, 378, 13–23. [Google Scholar] [CrossRef]
- Gatto, M.; Radu, C.M.; Luisetto, R.; Ghirardello, A.; Bonsembiante, F.; Trez, D.; Valentino, S.; Bottazzi, B.; Simioni, P.; Cavicchioli, L.; et al. Immunization with Pentraxin3 prevents transition from subclinical to clinical lupus nephritis in lupus-prone mice: Insights from renal ultrastructural findings. J. Autoimmun. 2020, 111, 102443. [Google Scholar] [CrossRef]
- Sjöwall, C.; Olin, A.I.; Skogh, T.; Wetterö, J.; Morgelin, M.; Nived, O.; Sturfelt, G.; Bengtsson, A.A. C-reactive protein, immunoglobulin G and complement co-localize in renal immune deposits of proliferative lupus nephritis. Autoimmunity 2013, 46, 205–214. [Google Scholar] [CrossRef]
- Li, Q.Y.; Li, H.Y.; Fu, G.; Yu, F.; Wu, Y.; Zhao, M.H. Autoantibodies against C-Reactive Protein Influence Complement Activation and Clinical Course in Lupus Nephritis. J. Am. Soc. Nephrol. 2017, 28, 3044–3054. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Hao, J.B.; Chu, H.; Wang, F.M.; Song, D.; Zhu, L.; Yu, F.; Li, Y.Z.; Song, Y.; Zhao, M.H. Genetic variants in FH are associated with renal histopathologic subtypes of lupus nephritis: A large cohort study from China. Lupus 2017, 26, 1309–1317. [Google Scholar] [CrossRef]
- Wang, F.M.; Song, D.; Pang, Y.; Song, Y.; Yu, F.; Zhao, M.H. The dysfunctions of complement factor H in lupus nephritis. Lupus 2016, 25, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Karpati, E.; Papp, A.; Schneider, A.E.; Hajnal, D.; Cserhalmi, M.; Csincsi, A.I.; Uzonyi, B.; Jozsi, M. Interaction of the Factor H Family Proteins FHR-1 and FHR-5 With DNA and Dead Cells: Implications for the Regulation of Complement Activation and Opsonization. Front Immunol. 2020, 11, 1297. [Google Scholar] [CrossRef] [PubMed]
- Hebecker, M.; Okemefuna, A.I.; Perkins, S.J.; Mihlan, M.; Huber-Lang, M.; Jozsi, M. Molecular basis of C-reactive protein binding and modulation of complement activation by factor H-related protein 4. Mol. Immunol. 2010, 47, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Mihlan, M.; Hebecker, M.; Dahse, H.M.; Halbich, S.; Huber-Lang, M.; Dahse, R.; Zipfel, P.F.; Jozsi, M. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol. Immunol. 2009, 46, 335–344. [Google Scholar] [CrossRef]
- Mihlan, M.; Blom, A.M.; Kupreishvili, K.; Lauer, N.; Stelzner, K.; Bergström, F.; Niessen, H.W.; Zipfel, P.F. Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J. 2011, 25, 4198–4210. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, A.P.; Trouw, L.A.; McGrath, F.D.; Hack, C.E.; Blom, A.M. Regulation of complement activation by C-reactive protein: Targeting of the inhibitory activity of C4b-binding protein. J. Immunol. 2006, 176, 7612–7620. [Google Scholar] [CrossRef]
- Sjöwall, C.; Wetterö, J.; Bengtsson, T.; Askendal, A.; Almroth, G.; Skogh, T.; Tengvall, P. Solid-phase classical complement activation by C-reactive protein (CRP) is inhibited by fluid-phase CRP-C1q interaction. Biochem. Biophys. Res. Commun. 2007, 352, 251–258. [Google Scholar] [CrossRef][Green Version]
- Roy, N.; Ohtani, K.; Matsuda, Y.; Mori, K.; Hwang, I.; Suzuki, Y.; Inoue, N.; Wakamiya, N. Collectin CL-P1 utilizes C-reactive protein for complement activation. Biochim. Biophys Acta. 2016, 1860, 1118–1128. [Google Scholar] [CrossRef]
- Lu, J.; Mold, C.; Du Clos, T.W.; Sun, P.D. Pentraxins and Fc Receptor-Mediated Immune Responses. Front Immunol. 2018, 9, 2607. [Google Scholar] [CrossRef]
- Bharadwaj, D.; Stein, M.P.; Volzer, M.; Mold, C.; Du Clos, T.W. The major receptor for C-reactive protein on leukocytes is fcgamma receptor II. J. Exp. Med. 1999, 190, 585–590. [Google Scholar] [CrossRef]
- Manolov, D.E.; Rocker, C.; Hombach, V.; Nienhaus, G.U.; Torzewski, J. Ultrasensitive confocal fluorescence microscopy of C-reactive protein interacting with FcgammaRIIa. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Mold, C.; Du Clos, T.W. C-reactive protein increases cytokine responses to Streptococcus pneumoniae through interactions with Fc gamma receptors. J. Immunol. 2006, 176, 7598–7604. [Google Scholar] [CrossRef] [PubMed]
- Båve, U.; Magnusson, M.; Eloranta, M.L.; Perers, A.; Alm, G.V.; Rönnblom, L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J. Immunol. 2003, 171, 3296–3302. [Google Scholar] [CrossRef] [PubMed]
- Mold, C.; Clos, T.W. C-reactive protein inhibits plasmacytoid dendritic cell interferon responses to autoantibody immune complexes. Arthritis. Rheum. 2013, 65, 1891–1901. [Google Scholar] [CrossRef]
- Lu, J.; Marjon, K.D.; Marnell, L.L.; Wang, R.; Mold, C.; Du Clos, T.W.; Sun, P. Recognition and functional activation of the human IgA receptor (FcalphaRI) by C-reactive protein. Proc. Natl. Acad. Sci. USA 2011, 108, 4974–4979. [Google Scholar] [CrossRef]
- Calabro, P.; Willerson, J.T.; Yeh, E.T. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation 2003, 108, 1930–1932. [Google Scholar] [CrossRef]
- Gould, J.M.; Weiser, J.N. Expression of C-reactive protein in the human respiratory tract. Infect. Immun. 2001, 69, 1747–1754. [Google Scholar] [CrossRef]
- Hernandez-Caldera, A.; Vernal, R.; Paredes, R.; Veloso-Matta, P.; Astorga, J.; Hernandez, M. Human periodontal ligament fibroblasts synthesize C-reactive protein and Th-related cytokines in response to interleukin (IL)-6 trans-signalling. Int. Endod. J. 2018, 51, 632–640. [Google Scholar] [CrossRef]
- Jabs, W.J.; Logering, B.A.; Gerke, P.; Kreft, B.; Wolber, E.M.; Klinger, M.H.; Fricke, L.; Steinhoff, J. The kidney as a second site of human C-reactive protein formation in vivo. Eur. J. Immunol. 2003, 33, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, J.; Han, C.; Wang, B.; Pang, X.; Mao, J. Angiotensin II induces the expression of c-reactive protein via MAPK-dependent signal pathway in U937 macrophages. Cell Physiol. Biochem. 2011, 27, 63–70. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, S.L.; Rzewnicki, D.; Samols, D.; Kushner, I. The effect of interleukin-1 on C-reactive protein expression in Hep3B cells is exerted at the transcriptional level. Biochem. J. 1995, 310, 143–148. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J. Biol. Chem. 1996, 271, 9503–9509. [Google Scholar] [CrossRef]
- Agrawal, A.; Cha-Molstad, H.; Samols, D.; Kushner, I. Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein beta (C/EBP beta) and Rel p50. J. Immunol. 2001, 166, 2378–2384. [Google Scholar] [CrossRef] [PubMed]
- Enocsson, H.; Sjöwall, C.; Skogh, T.; Eloranta, M.L.; Rönnblom, L.; Wetterö, J. Interferon-alpha mediates suppression of C-reactive protein: Explanation for muted C-reactive protein response in lupus flares? Arthritis. Rheum. 2009, 60, 3755–3760. [Google Scholar] [CrossRef]
- Gabay, C.; Genin, B.; Mentha, G.; Iynedjian, P.B.; Roux-Lombard, P.; Guerne, P.A. IL-1 receptor antagonist (IL-1Ra) does not inhibit the production of C-reactive protein or serum amyloid A protein by human primary hepatocytes. Differential regulation in normal and tumour cells. Clin. Exp. Immunol. 1995, 100, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Lanham, J.G.; De Beer, F.C. C-reactive protein in SLE. Clin Rheum Dis 1982, 8, 91–103. [Google Scholar] [CrossRef]
- Rönnblom, L.; Eloranta, M.L. The interferon signature in autoimmune diseases. Curr. Opin. Rheumatol. 2013, 25, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Sonoda, S.; Urano, T.; Yamada, T.; Okada, M. Monitoring both serum amyloid protein A and C-reactive protein as inflammatory markers in infectious diseases. Clin. Chem. 1993, 39, 293–297. [Google Scholar] [CrossRef]
- Eloranta, M.L.; Rönnblom, L. Cause and consequences of the activated type I interferon system in SLE. J. Mol. Med. 2016, 94, 1103–1110. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors--distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Enocsson, H.; Sjöwall, C.; Kastbom, A.; Skogh, T.; Eloranta, M.L.; Rönnblom, L.; Wetterö, J. Association of serum C-reactive protein levels with lupus disease activity in the absence of measurable interferon-alpha and a C-reactive protein gene variant. Arthritis. Rheumatol. 2014, 66, 1568–1573. [Google Scholar] [CrossRef]
- Hage, F.G.; Szalai, A.J. C-reactive protein gene polymorphisms, C-reactive protein blood levels, and cardiovascular disease risk. J. Am. Coll. Cardiol. 2007, 50, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Szalai, A.J.; Alarcon, G.S.; Calvo-Alen, J.; Toloza, S.M.; McCrory, M.A.; Edberg, J.C.; McGwin, G., Jr.; Bastian, H.M.; Fessler, B.J.; Vila, L.M.; et al. Systemic lupus erythematosus in a multiethnic US Cohort (LUMINA). XXX: Association between C-reactive protein (CRP) gene polymorphisms and vascular events. Rheumatology 2005, 44, 864–868. [Google Scholar] [CrossRef]
- Szalai, A.J.; Wu, J.; Lange, E.M.; McCrory, M.A.; Langefeld, C.D.; Williams, A.; Zakharkin, S.O.; George, V.; Allison, D.B.; Cooper, G.S.; et al. Single-nucleotide polymorphisms in the C-reactive protein (CRP) gene promoter that affect transcription factor binding, alter transcriptional activity, and associate with differences in baseline serum CRP level. J. Mol. Med. 2005, 83, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Enocsson, H.; Gullstrand, B.; Eloranta, M.L.; Wetterö, J.; Leonard, D.; Rönnblom, L.; Bengtsson, A.A.; Sjöwall, C. C-Reactive Protein Levels in Systemic Lupus Erythematosus Are Modulated by the Interferon Gene Signature and CRP Gene Polymorphism rs1205. Front. Immunol. 2021, 11, 622326. [Google Scholar] [CrossRef]
- Potempa, L.A.; Maldonado, B.A.; Laurent, P.; Zemel, E.S.; Gewurz, H. Antigenic, electrophoretic and binding alterations of human C-reactive protein modified selectively in the absence of calcium. Mol. Immunol. 1983, 20, 1165–1175. [Google Scholar] [CrossRef]
- Verma, S.; Szmitko, P.E.; Yeh, E.T. C-reactive protein: Structure affects function. Circulation 2004, 109, 1914–1917. [Google Scholar] [CrossRef]
- Ji, S.R.; Wu, Y.; Zhu, L.; Potempa, L.A.; Sheng, F.L.; Lu, W.; Zhao, J. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRP(m). FASEB J. 2007, 21, 284–294. [Google Scholar] [CrossRef]
- Potempa, L.A.; Siegel, J.N.; Fiedel, B.A.; Potempa, R.T.; Gewurz, H. Expression, detection and assay of a neoantigen (Neo-CRP) associated with a free, human C-reactive protein subunit. Mol. Immunol. 1987, 24, 531–541. [Google Scholar] [CrossRef]
- Schwedler, S.B.; Filep, J.G.; Galle, J.; Wanner, C.; Potempa, L.A. C-reactive protein: A family of proteins to regulate cardiovascular function. Am. J. Kidney. Dis. 2006, 47, 212–222. [Google Scholar] [CrossRef]
- Skogh, T.; Stendahl, O. Complement-mediated delay in immune complex clearance from the blood owing to reduced deposition outside the reticuloendothelial system. Immunology 1983, 49, 53–59. [Google Scholar]
- Motie, M.; Brockmeier, S.; Potempa, L.A. Binding of model soluble immune complexes to modified C-reactive protein. J. Immunol. 1996, 156, 4435–4441. [Google Scholar] [PubMed]
- Eisenhardt, S.U.; Habersberger, J.; Murphy, A.; Chen, Y.C.; Woollard, K.J.; Bassler, N.; Qian, H.; von Zur Muhlen, C.; Hagemeyer, C.E.; Ahrens, I.; et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ. Res. 2009, 105, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Liu, X.L.; Liu, Y.T.; Jia, Z.K.; Filep, J.G.; Potempa, L.A.; Ji, S.R.; Wu, Y. Matrix sieving-enforced retrograde transcytosis regulates tissue accumulation of C-reactive protein. Cardiovasc Res. 2019, 115, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Iso, H.; Cui, R.; Date, C.; Kikuchi, S.; Tamakoshi, A.; Group, J.S. C-reactive protein levels and risk of mortality from cardiovascular disease in Japanese: The JACC Study. Atherosclerosis 2009, 207, 291–297. [Google Scholar] [CrossRef]
- Ji, S.R.; Wu, Y.; Potempa, L.A.; Qiu, Q.; Zhao, J. Interactions of C-reactive protein with low-density lipoproteins: Implications for an active role of modified C-reactive protein in atherosclerosis. Int. J. Biochem. Cell Biol. 2006, 38, 648–661. [Google Scholar] [CrossRef]
- Jiang, S.; Bao, Y.; Hou, X.; Fang, Q.; Wang, C.; Pan, J.; Zuo, Y.; Zhong, W.; Xiang, K.; Jia, W. Serum C-reactive protein and risk of cardiovascular events in middle-aged and older chinese population. Am. J. Cardiol. 2009, 103, 1727–1731. [Google Scholar] [CrossRef]
- Molins, B.; Pena, E.; Vilahur, G.; Mendieta, C.; Slevin, M.; Badimon, L. C-reactive protein isoforms differ in their effects on thrombus growth. Arterioscler. Thromb Vasc. Biol. 2008, 28, 2239–2246. [Google Scholar] [CrossRef]
- Ridker, P.M.; Paynter, N.P.; Rifai, N.; Gaziano, J.M.; Cook, N.R. C-reactive protein and parental history improve global cardiovascular risk prediction: The Reynolds Risk Score for men. Circulation 2008, 118, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Habersberger, J.; Strang, F.; Scheichl, A.; Htun, N.; Bassler, N.; Merivirta, R.M.; Diehl, P.; Krippner, G.; Meikle, P.; Eisenhardt, S.U.; et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc Res. 2012, 96, 64–72. [Google Scholar] [CrossRef]
- Strang, F.; Scheichl, A.; Chen, Y.C.; Wang, X.; Htun, N.M.; Bassler, N.; Eisenhardt, S.U.; Habersberger, J.; Peter, K. Amyloid plaques dissociate pentameric to monomeric C-reactive protein: A novel pathomechanism driving cortical inflammation in Alzheimer’s disease? Brain Pathol. 2012, 22, 337–346. [Google Scholar] [CrossRef]
- Eisenhardt, S.U.; Thiele, J.R.; Bannasch, H.; Stark, G.B.; Peter, K. C-reactive protein: How conformational changes influence inflammatory properties. Cell Cycle. 2009, 8, 3885–3892. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.R.; Ma, L.; Bai, C.J.; Shi, J.M.; Li, H.Y.; Potempa, L.A.; Filep, J.G.; Zhao, J.; Wu, Y. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J. 2009, 23, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Khreiss, T.; Jozsef, L.; Hossain, S.; Chan, J.S.; Potempa, L.A.; Filep, J.G. Loss of pentameric symmetry of C-reactive protein is associated with delayed apoptosis of human neutrophils. J. Biol. Chem. 2002, 277, 40775–40781. [Google Scholar] [CrossRef] [PubMed]
- Khreiss, T.; Jozsef, L.; Potempa, L.A.; Filep, J.G. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation 2004, 109, 2016–2022. [Google Scholar] [CrossRef] [PubMed]
- Khreiss, T.; Jozsef, L.; Potempa, L.A.; Filep, J.G. Opposing effects of C-reactive protein isoforms on shear-induced neutrophil-platelet adhesion and neutrophil aggregation in whole blood. Circulation 2004, 110, 2713–2720. [Google Scholar] [CrossRef]
- Khreiss, T.; Jozsef, L.; Potempa, L.A.; Filep, J.G. Loss of pentameric symmetry in C-reactive protein induces interleukin-8 secretion through peroxynitrite signaling in human neutrophils. Circ. Res. 2005, 97, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Lauer, N.; Mihlan, M.; Hartmann, A.; Schlotzer-Schrehardt, U.; Keilhauer, C.; Scholl, H.P.; Charbel Issa, P.; Holz, F.; Weber, B.H.; Skerka, C.; et al. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J. Immunol. 2011, 187, 4374–4383. [Google Scholar] [CrossRef]
- Molins, B.; Pena, E.; de la Torre, R.; Badimon, L. Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc. Res. 2011, 92, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Ji, S.R.; Bai, C.J.; El Kebir, D.; Li, H.Y.; Shi, J.M.; Zhu, W.; Costantino, S.; Zhou, H.H.; Potempa, L.A.; et al. A redox switch in C-reactive protein modulates activation of endothelial cells. FASEB J. 2011, 25, 3186–3196. [Google Scholar] [CrossRef] [PubMed]
- Zouki, C.; Haas, B.; Chan, J.S.; Potempa, L.A.; Filep, J.G. Loss of pentameric symmetry of C-reactive protein is associated with promotion of neutrophil-endothelial cell adhesion. J. Immunol. 2001, 167, 5355–5361. [Google Scholar] [CrossRef] [PubMed]
- Boncler, M.; Kehrel, B.; Szewczyk, R.; Stec-Martyna, E.; Bednarek, R.; Brodde, M.; Watala, C. Oxidation of C-reactive protein by hypochlorous acid leads to the formation of potent platelet activator. Int. J. Biol. Macromol. 2018, 107, 2701–2714. [Google Scholar] [CrossRef]
- Schwedler, S.B.; Guderian, F.; Dammrich, J.; Potempa, L.A.; Wanner, C. Tubular staining of modified C-reactive protein in diabetic chronic kidney disease. Nephrol. Dial. Transplant 2003, 18, 2300–2307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Slevin, M.; Matou-Nasri, S.; Turu, M.; Luque, A.; Rovira, N.; Badimon, L.; Boluda, S.; Potempa, L.; Sanfeliu, C.; de Vera, N.; et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol. 2010, 20, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Robey, F.A.; Jones, K.D.; Steinberg, A.D. C-reactive protein mediates the solubilization of nuclear DNA by complement in vitro. J. Exp. Med. 1985, 161, 1344–1356. [Google Scholar] [CrossRef]
- Bell, S.A.; Du Clos, T.W.; Khursigara, G.; Picazo, J.J.; Rubin, R.L. Autoantibodies to cryptic epitopes of C-reactive protein and other acute phase proteins in the toxic oil syndrome. J. Autoimmun. 1995, 8, 293–303. [Google Scholar] [CrossRef]
- Bell, S.A.; Faust, H.; Schmid, A.; Meurer, M. Autoantibodies to C-reactive protein (CRP) and other acute-phase proteins in systemic autoimmune diseases. Clin. Exp. Immunol. 1998, 113, 327–332. [Google Scholar] [CrossRef]
- Sjöwall, C.; Eriksson, P.; Almer, S.; Skogh, T. Autoantibodies to C-reactive protein is a common finding in SLE, but not in primary Sjogren’s syndrome, rheumatoid arthritis or inflammatory bowel disease. J. Autoimmun. 2002, 19, 155–160. [Google Scholar] [CrossRef]
- Sjöwall, C.; Bengtsson, A.A.; Sturfelt, G.; Skogh, T. Serum levels of autoantibodies against monomeric C-reactive protein are correlated with disease activity in systemic lupus erythematosus. Arthritis. Res. Ther. 2004, 6, R87–R94. [Google Scholar] [CrossRef]
- Rosenau, B.J.; Schur, P.H. Antibodies to C reactive protein. Ann. Rheum. Dis. 2006, 65, 674–676. [Google Scholar] [CrossRef]
- Sjöwall, C.; Cardell, K.; Boström, E.A.; Bokarewa, M.I.; Enocsson, H.; Ekstedt, M.; Lindvall, L.; Fryden, A.; Almer, S. High prevalence of autoantibodies to C-reactive protein in patients with chronic hepatitis C infection: Association with liver fibrosis and portal inflammation. Hum. Immunol. 2012, 73, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Jakuszko, K.; Krajewska, M.; Halon, A.; Koscielska-Kasprzak, K.; Myszka, M.; Zabinska, M.; Augustyniak-Bartosik, H.; Rukasz, D.; Weyde, W.; Klinger, M. Pathogenic role of antibodies against monomeric C-reactive protein in tubulointerstitial nephritis and uveitis syndrome. Intern. Med. J. 2014, 44, 809–812. [Google Scholar] [CrossRef]
- Figueredo, M.A.; Rodriguez, A.; Ruiz-Yague, M.; Romero, M.; Fernandez-Cruz, A.; Gomez-de la Concha, E.; Patino, R. Autoantibodies against C-reactive protein: Clinical associations in systemic lupus erythematosus and primary antiphospholipid syndrome. J. Rheumatol. 2006, 33, 1980–1986. [Google Scholar] [PubMed]
- Pesickova, S.S.; Rysava, R.; Lenicek, M.; Vitek, L.; Potlukova, E.; Hruskova, Z.; Jancova, E.; Honsova, E.; Zavada, J.; Trendelenburg, M.; et al. Prognostic value of anti-CRP antibodies in lupus nephritis in long-term follow-up. Arthritis Res. Ther. 2015, 17, 371. [Google Scholar] [CrossRef]
- Mathsson, L.; Ahlin, E.; Sjöwall, C.; Skogh, T.; Rönnelid, J. Cytokine induction by circulating immune complexes and signs of in-vivo complement activation in systemic lupus erythematosus are associated with the occurrence of anti-Sjogren’s syndrome A antibodies. Clin. Exp. Immunol. 2007, 147, 513–520. [Google Scholar] [CrossRef]
- O’Neill, S.G.; Giles, I.; Lambrianides, A.; Manson, J.; D’Cruz, D.; Schrieber, L.; March, L.M.; Latchman, D.S.; Isenberg, D.A.; Rahman, A. Antibodies to apolipoprotein A-I, high-density lipoprotein, and C-reactive protein are associated with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Wener, M.H.; Uwatoko, S.; Mannik, M. Antibodies to the collagen-like region of C1q in sera of patients with autoimmune rheumatic diseases. Arthritis Rheum. 1989, 32, 544–551. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).