
Design and Synthesis of Hybrid Compounds as Epigenetic Modifiers

 ,
,  and
and 
Abstract
:
1. Introduction
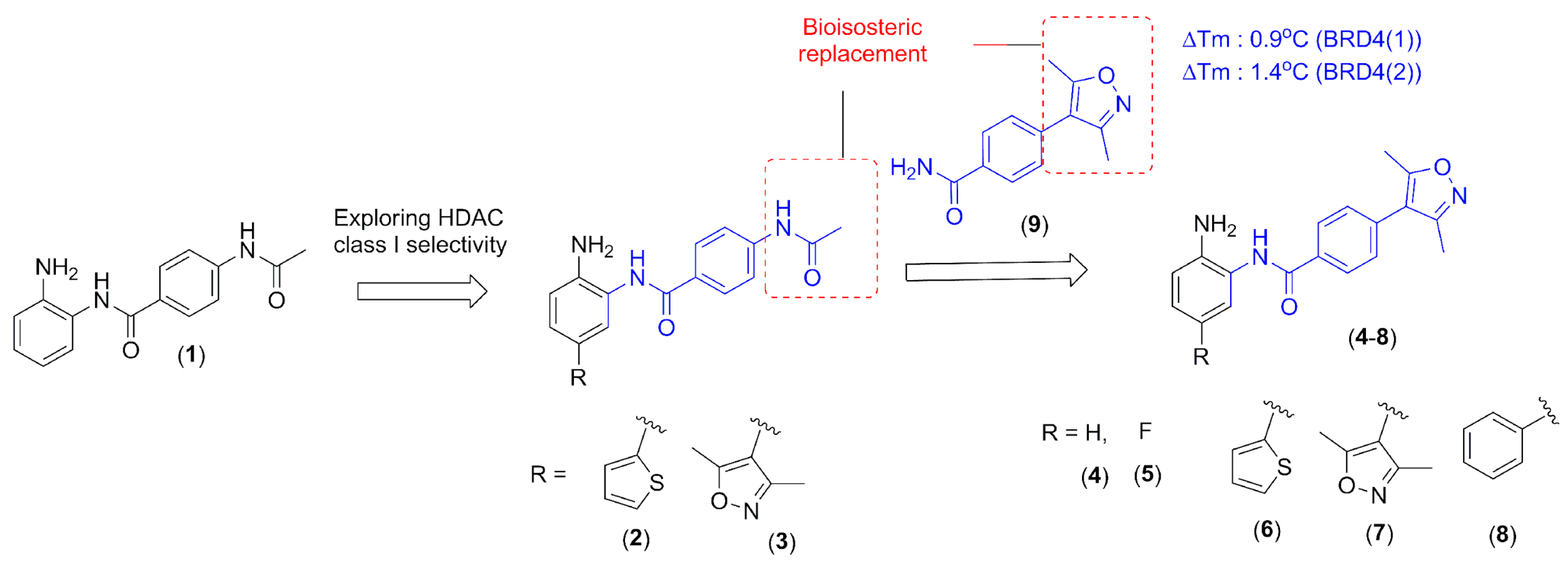
2. Results and Discussion
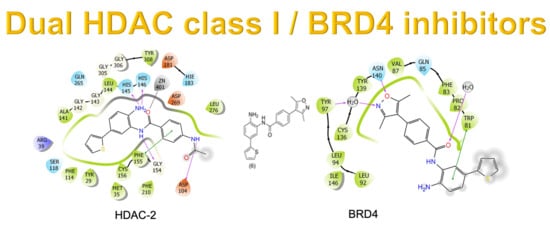
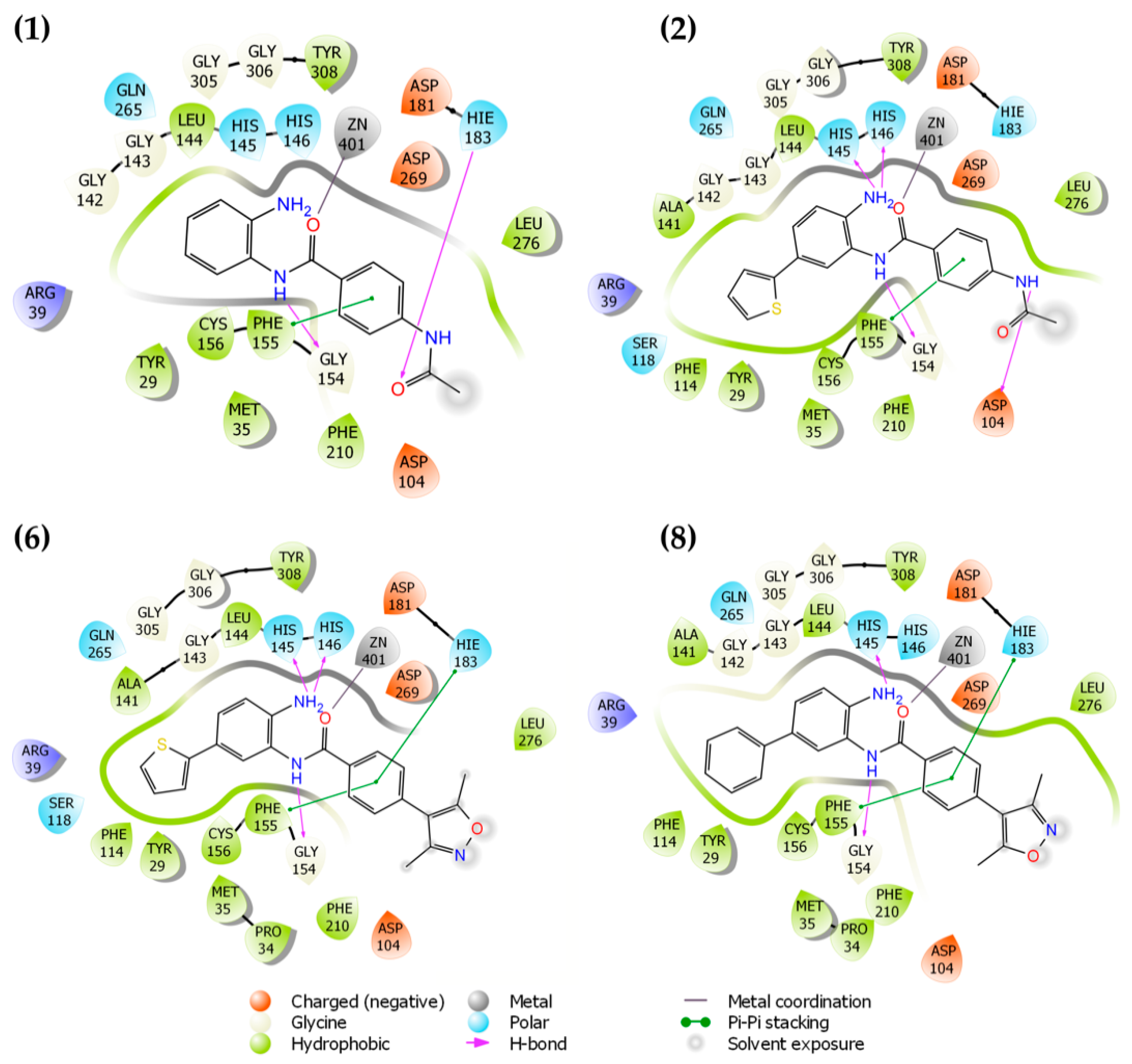
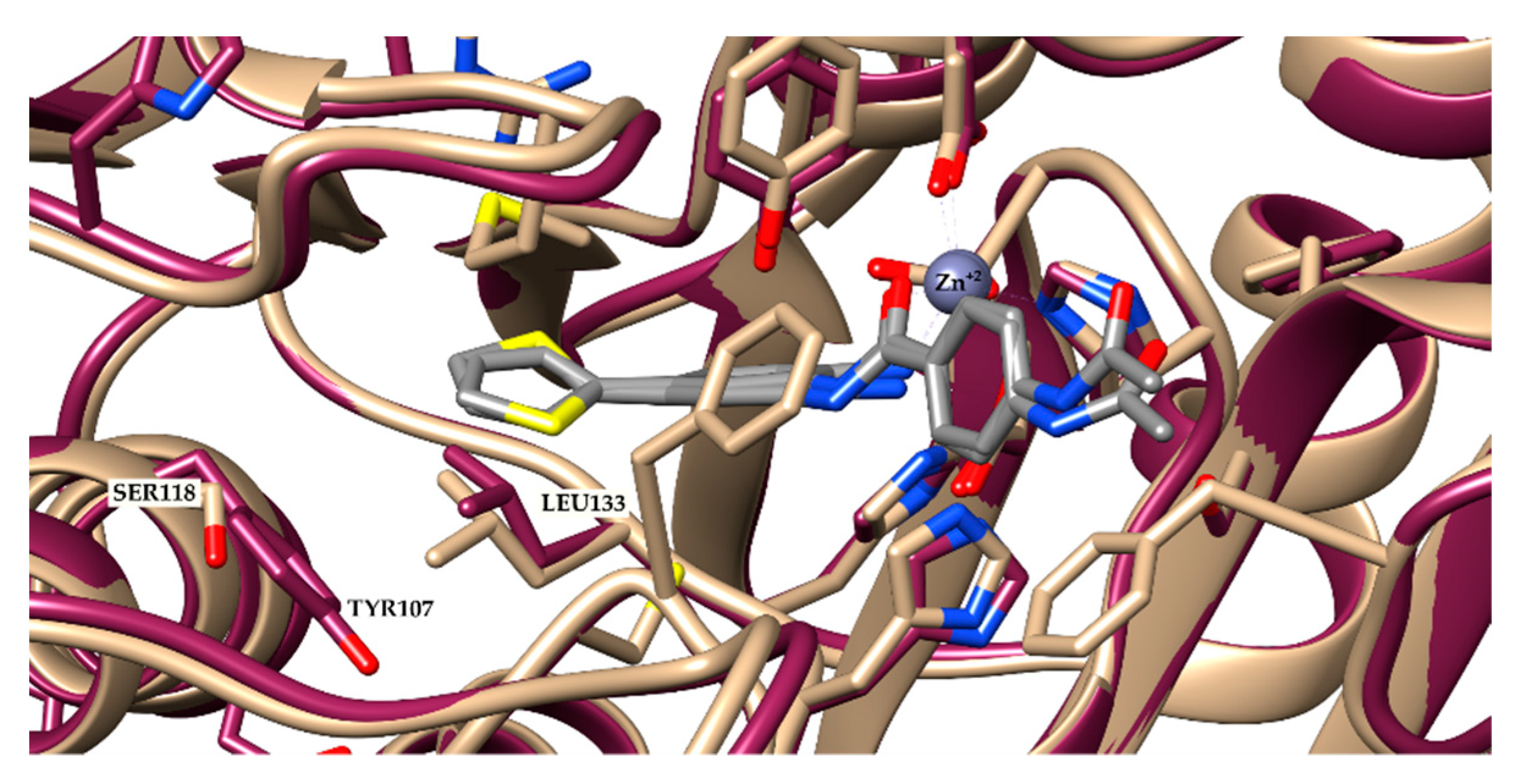
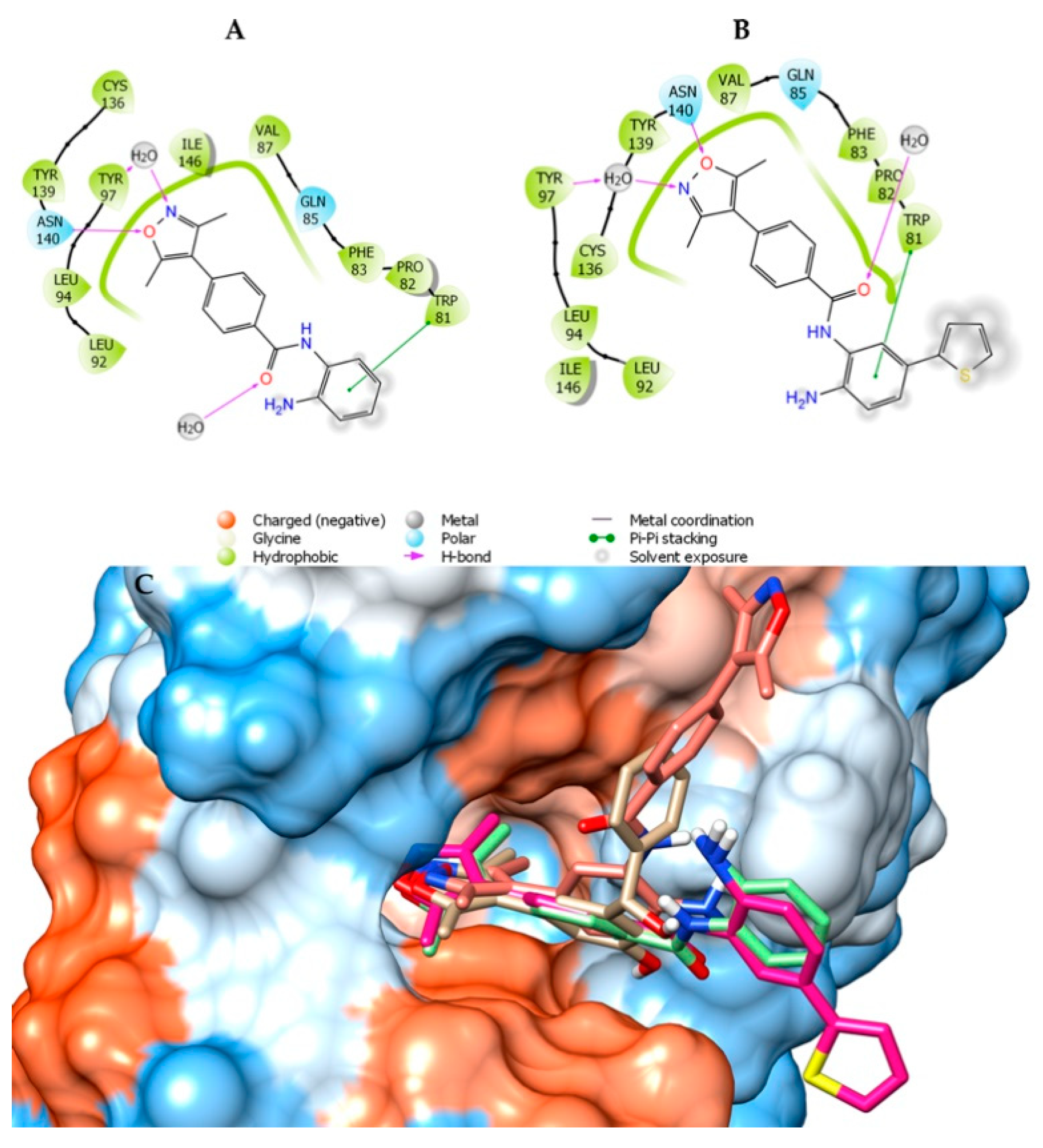
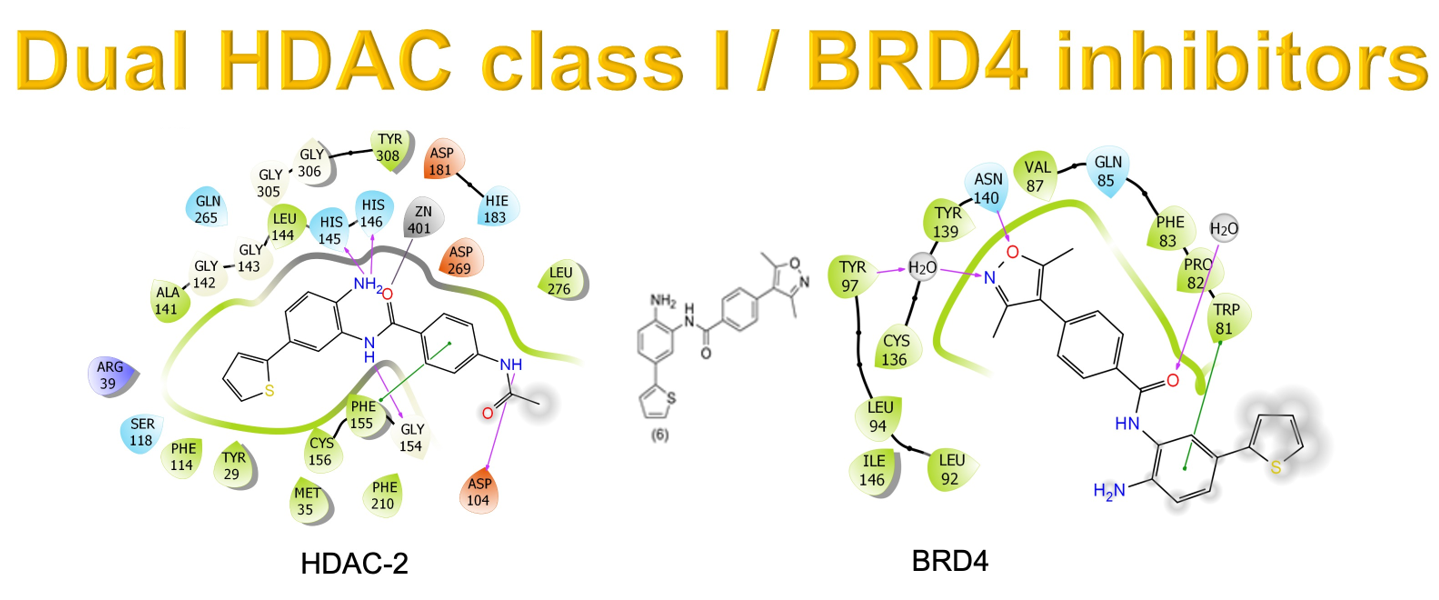
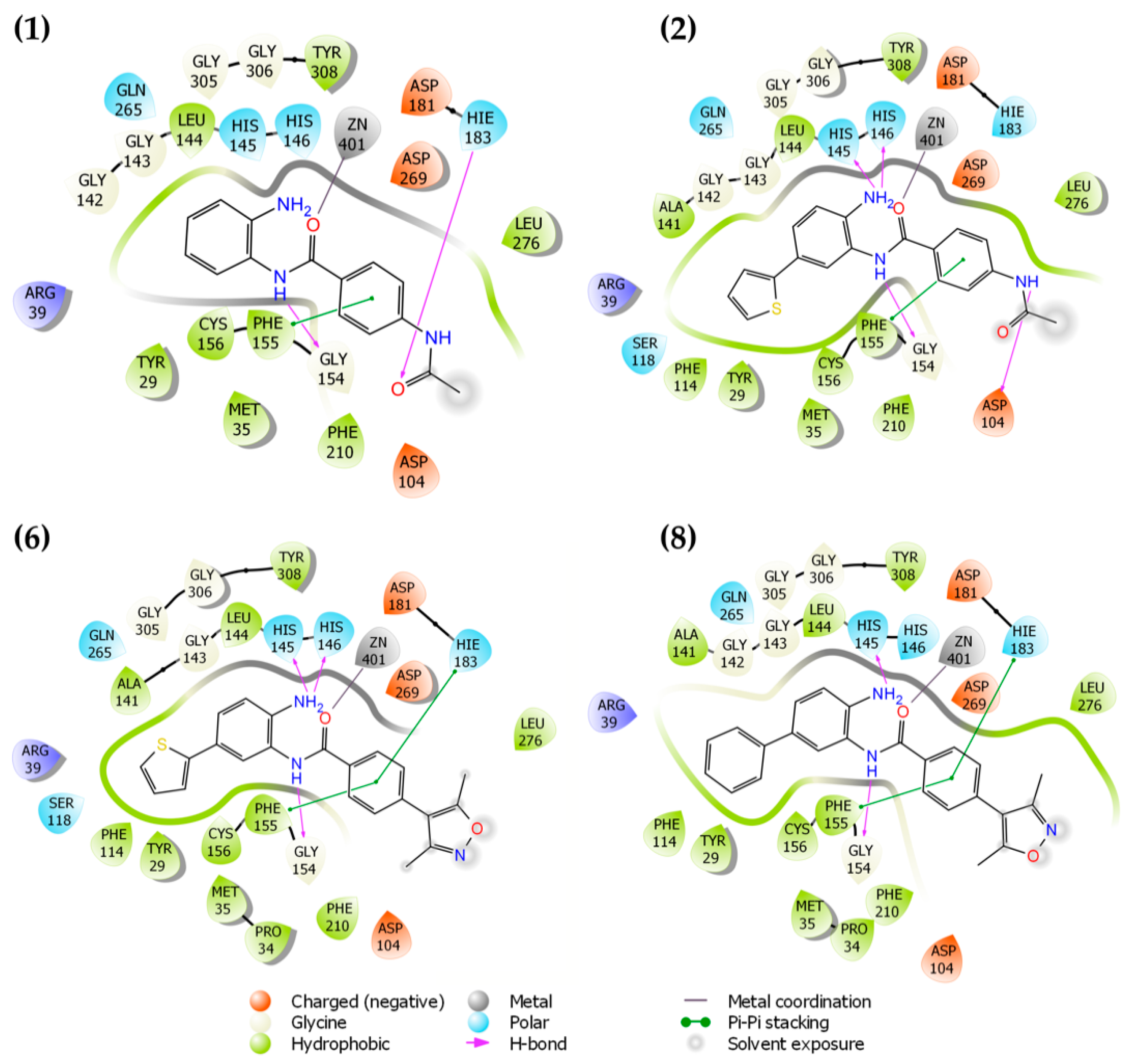
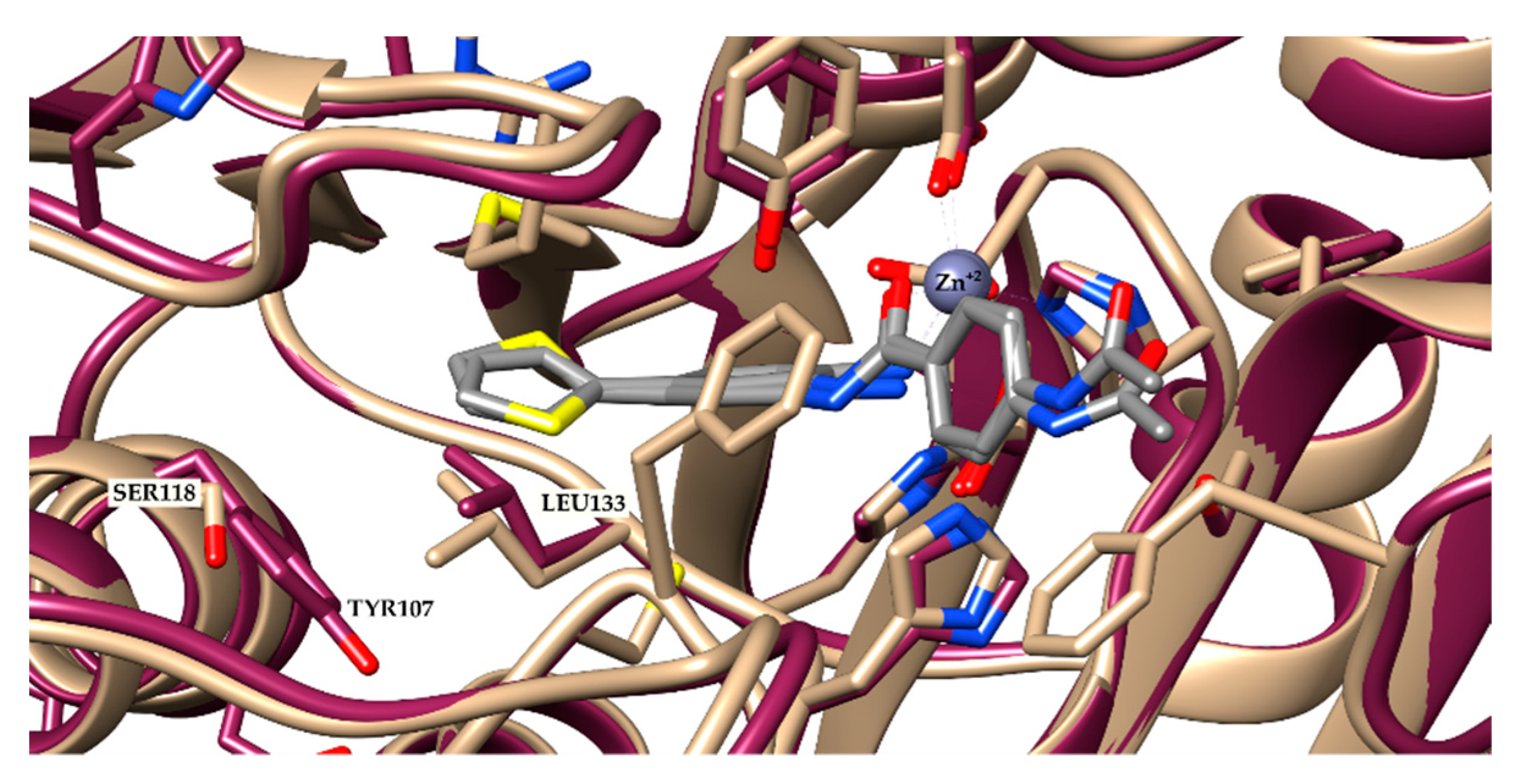
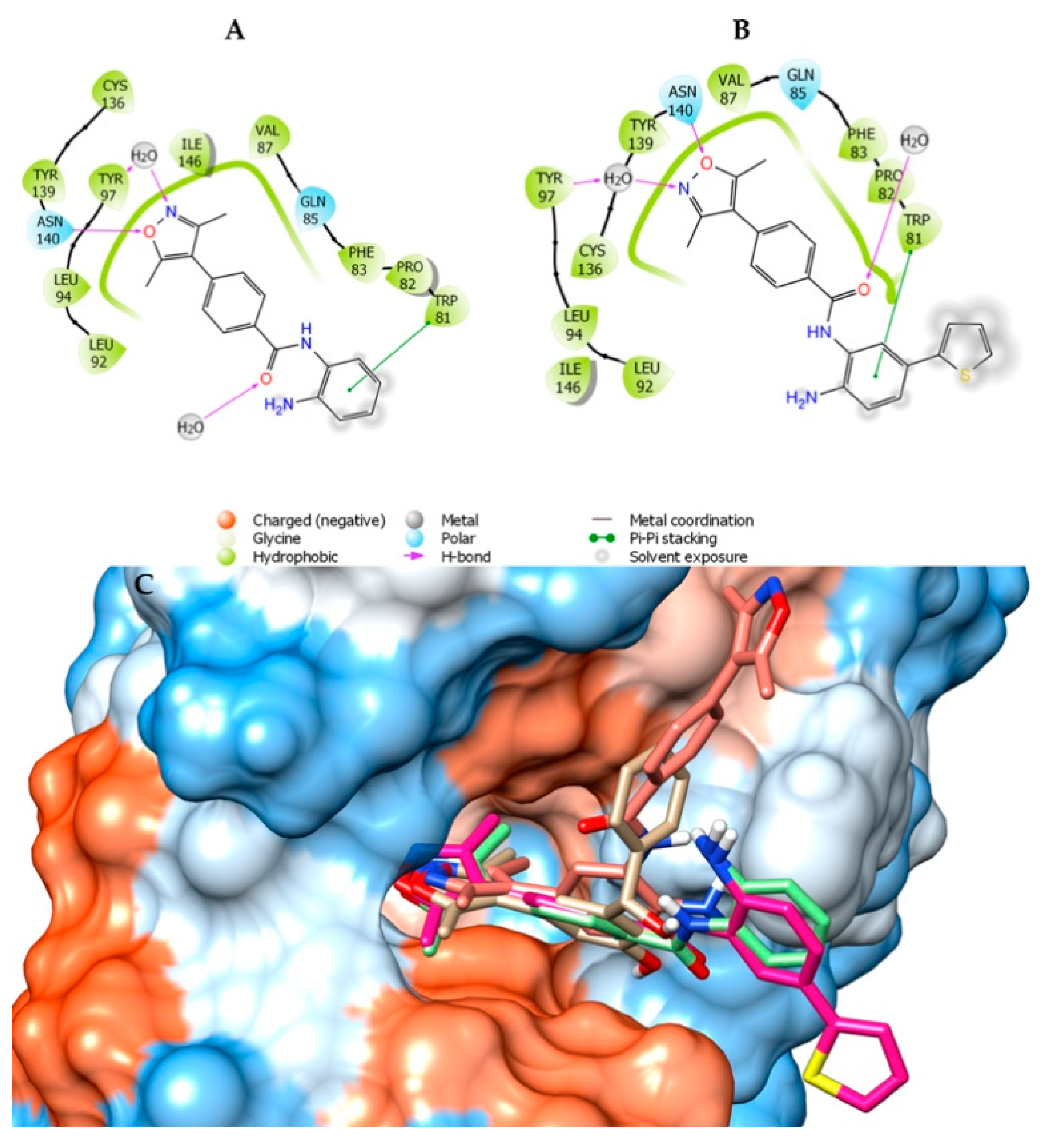
2.1. Molecular Docking and In Silico ADME Properties
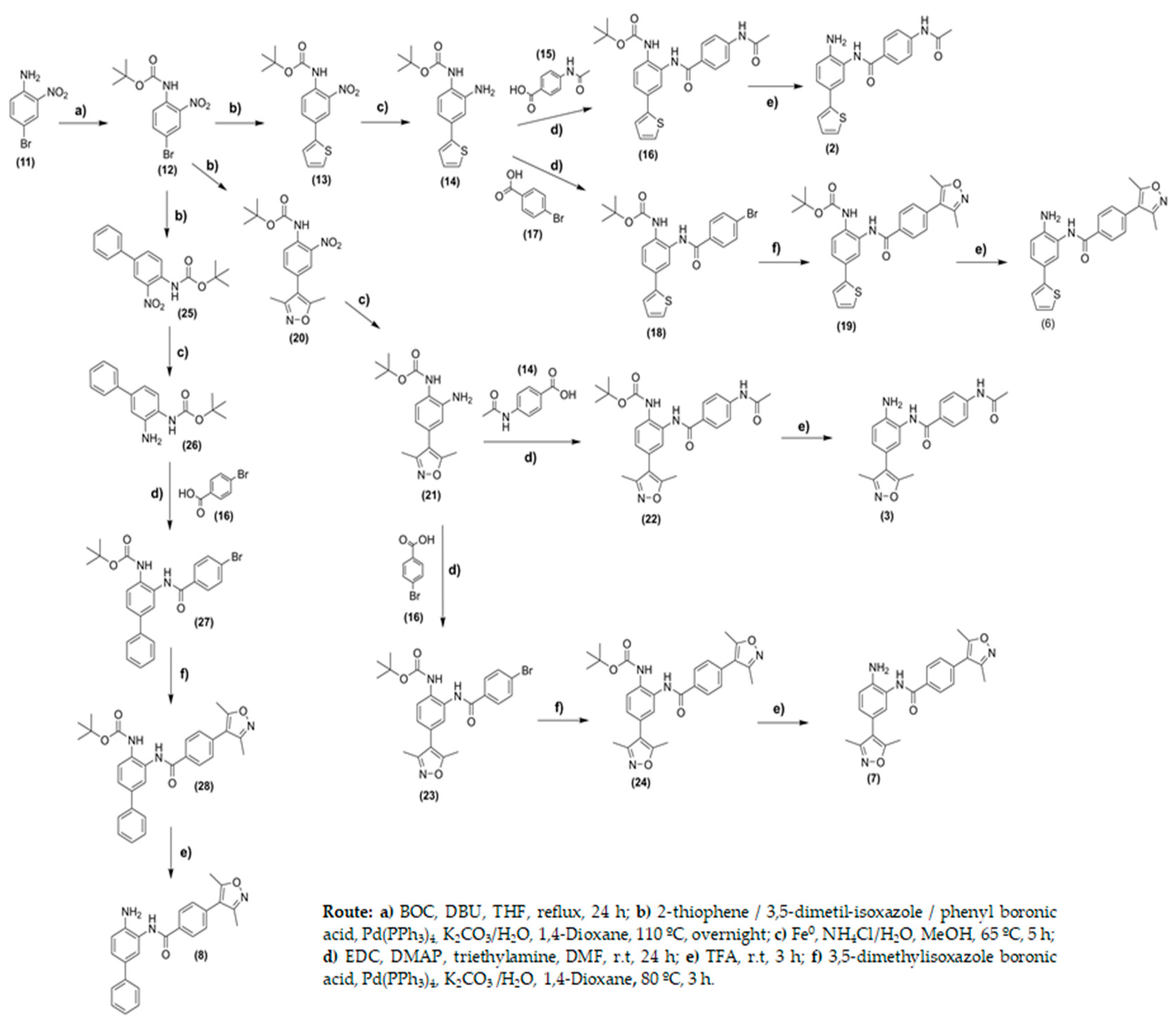
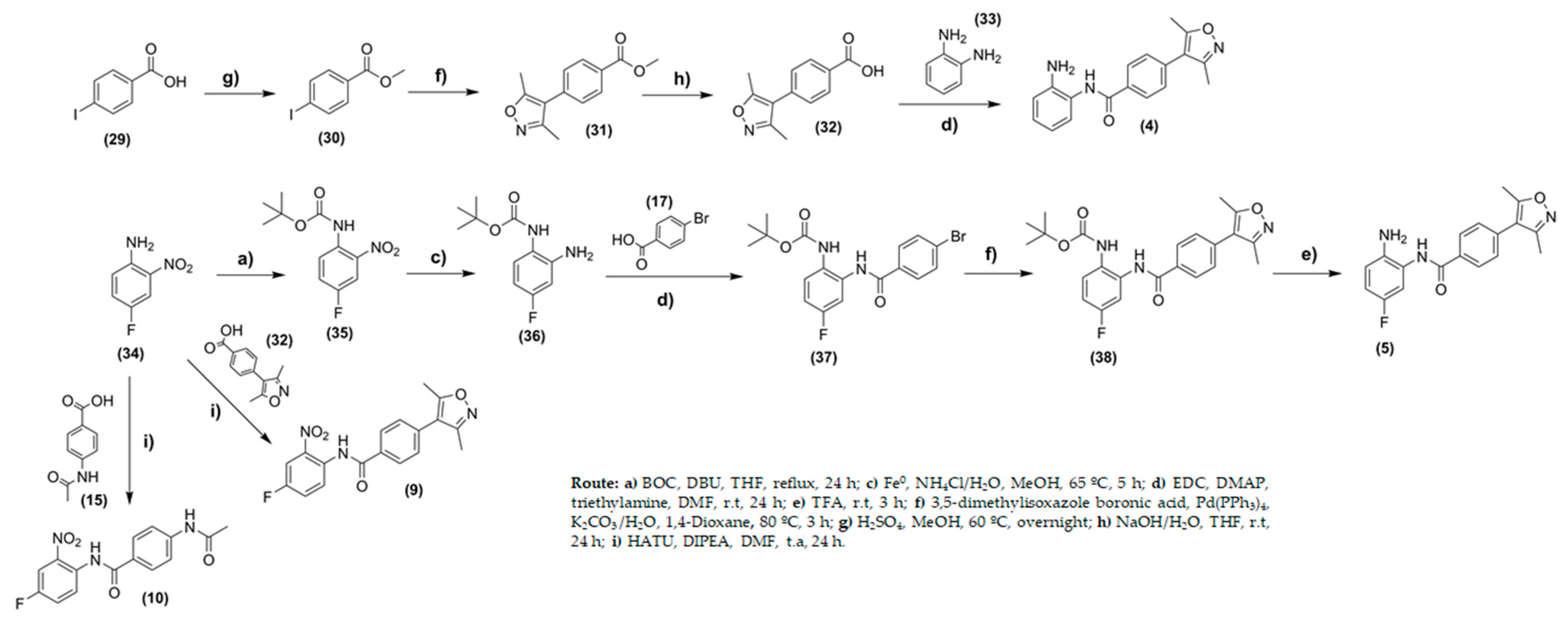
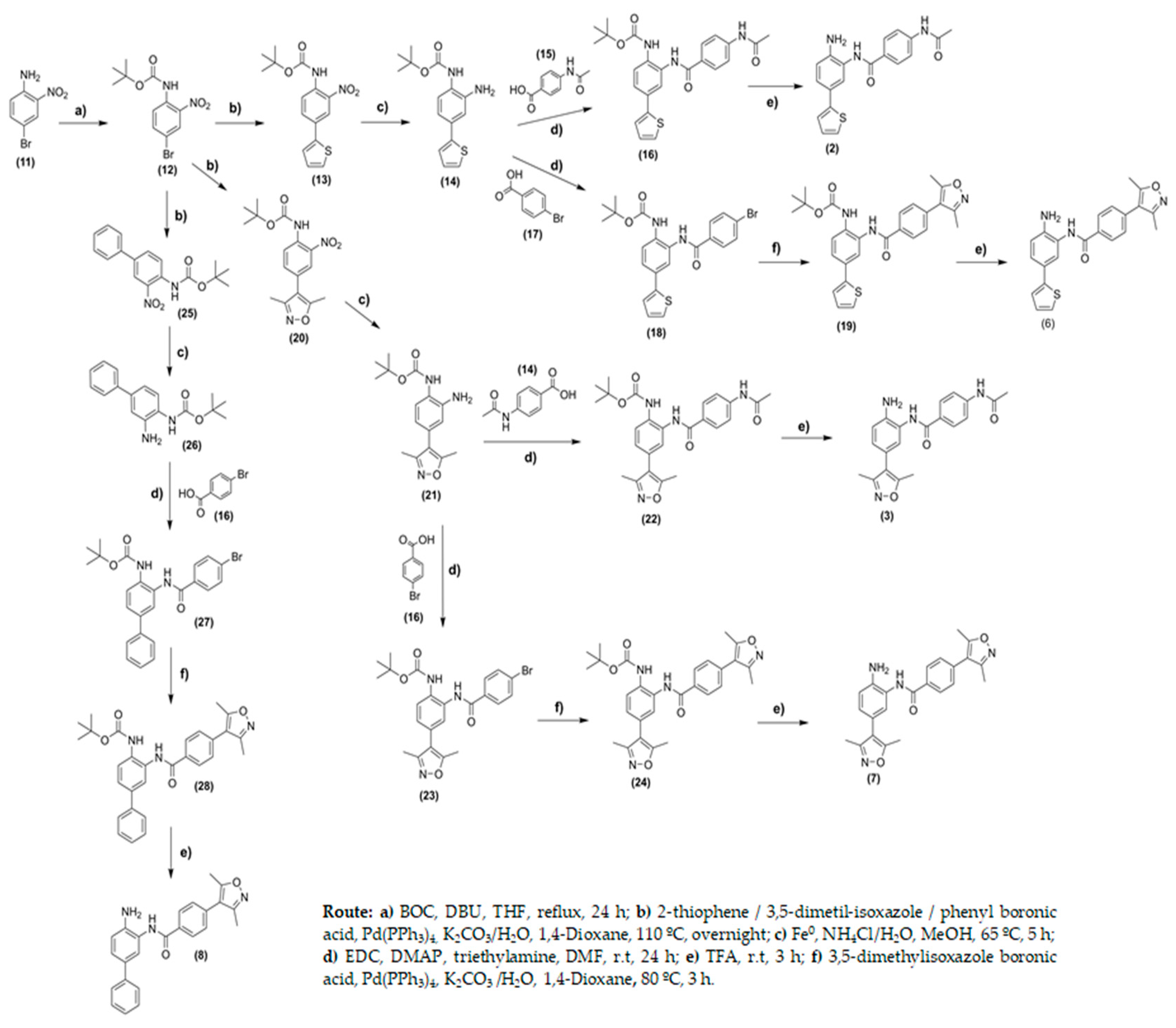
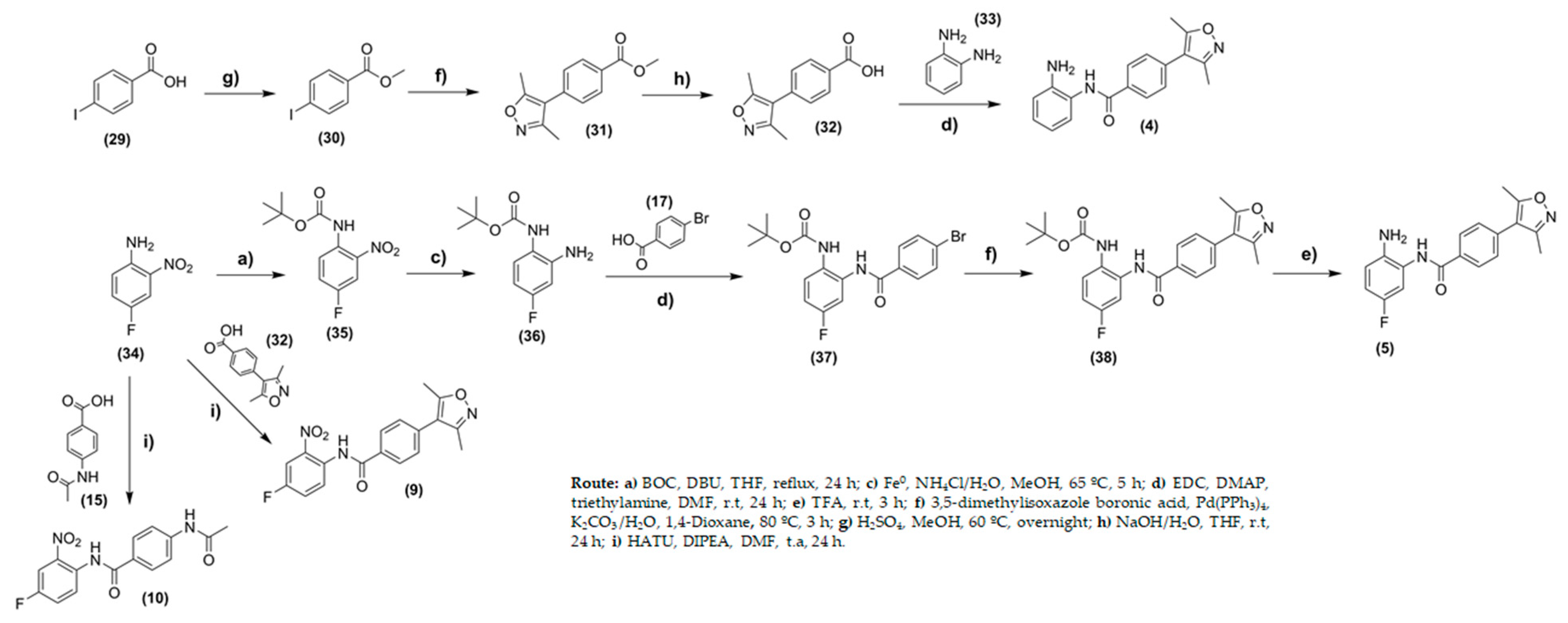
2.2. Chemistry
2.3. Enzymatic Inhibition Assays
3. Materials and Methods
3.1. Computational Methods
3.1.1. Molecular Docking
3.1.2. In Silico Prediction of ADME Properties
3.2. Chemistry
3.2.1. General
3.2.2. Synthesis of Tert-butyl (4-(3,5-dimethylisoxazol-4-yl)-2-nitrophenyl)carbamate (20)
3.2.3. General Procedures for the Synthesis of Tert-butyl (2-amino-4-phenyl)carbamate (21), (26) and (36)
Tert-butyl (2-amino-4-(3,5-dimethylisoxazol-4-yl)phenyl)carbamate (21)
Tert-butyl (2-amino-4-fluorophenyl)carbamate (36)
3.2.4. General Procedures for the Synthesis of 4-acetyl-N-(2-amino-5-phenyl)benzamide derivatives (2) and (3)
4-Acetyl-N-(2-amino-5-(thiophen-2-yl)phenyl)benzamide (2)
4-Acetyl-N-(2-amino-5-(3,5-dimethylisoxazol-4-yl)phenyl)benzamide (3)
3.2.5. Procedures for the Synthesis of N-(2-Aminophenyl)-4-(3,5-dimethylisoxazol-4-yl)benzamide (4)
Synthesis of 4-Methyl Iodine Benzoate (30)
Synthesis of Methyl 4-(3,5-Dimethylisoxazol-4-yl)benzoate (31)
Synthesis of 4-(3,5-Dimethylisoxazol-4-yl)benzoic Acid (32)
N-(2-Aminophenyl)-4-(3,5-dimethylisoxazol-4-yl)benzamide (4)
3.2.6. General Procedure for the Synthesis of Tert-butyl (2-(4-bromo-benzamide) Derivatives (18) and (23)
Tert-butyl (2-(4-bromobenzamide)-4-(thiophen-2-yl)phenyl)carbamate (18)
Tert-butyl(2-(4-bromobenzamide)-4-(3,5-dimethylisoxazol)4yl)phenyl)carbamate (23)
3.2.7. General Procedure for the Synthesis of Tert-butyl (2-(4-bromo-benzamide) Derivatives (27) and (37)
Tert-butyl (3-(4-bromobenzamido)-[1,1’-biphenyl]-4-yl)carbamate (27)
Tert-butyl (4-fluoro-2-(4-iodobenzamido)phenyl)carbamate (37)
3.2.8. General Procedure for the Synthesis of (5) (6), (7) and (8)
N-(2-Amino-5-fluorophenyl)-4-(3,5-dimethylisoxazol-4-yl)benzamide (5)
N-(2-Amino-5-(thiophen-2-yl)phenyl)-4-(3,5-dimethylisoxazol-4-yl)benzamide (6)
N-(2-Amino-5-(3,5-dimethylisoxazol-4-yl)phenyl)-4-(3,5-dimethylisoxazol-4-yl)benzamide (7)
N-(4-Amino-[1,1’-biphenyl]-3-yl)-4-(3,5-dimethyl-4,5-dihydroisoxazol-4-yl)benzamide (8)
3.2.9. General Procedure for the Synthesis of (9) and (10)
4-Acetamide-N-(4-fluoro-2-nitrophenyl)benzamide (9)
4-(3,5-Dimethylisoxazol-4-yl)-N-(4-fluoro-2-nitrophenyl)benzamide (10)
3.3. Enzymatic Inhibition Assays
3.3.1. HDAC Inhibition Assay
3.3.2. BRD4 Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [Green Version]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, G.N.; Rana, P.; Venkatesh, A.; Chatterjee, D.R.; Contractor, D.; Satpute, D.P.; Nagpure, M.; Jain, A.; Kumar, D. Paradigm shift of “classical” HDAC inhibitors to “hybrid” HDAC inhibitors in therapeutic interventions. Eur. J. Med. Chem. 2021, 209, 112844. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar]
- Bass, A.K.A.; El-Zoghbi, M.S.; Nageeb, E.-S.M.; Mohamed, M.F.A.; Bader, M.; Abuo-Rahma, G.E.-D.A. Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors. Eur. J. Med. Chem. 2021, 209, 112904. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Ghorbani, S.; Khanizadeh, S.; Namdari, H.; Faghihloo, E.; Akbari, A. Histone deacetylases in virus-associated cancers. Rev. Med. Virol. 2020, 30, e2085. [Google Scholar] [CrossRef]
- De Freitas, N.L.; Deberaldini, M.G.; Gomes, D.; Pavan, A.R.; Sousa, Â.; Dos Santos, J.L.; Soares, C.P. Histone Deacetylase Inhibitors as Therapeutic Interventions on Cervical Cancer Induced by Human Papillomavirus. Front. Cell Dev. Biol. 2021, 8, 592868. [Google Scholar] [CrossRef]
- Herbein, G.; Wendling, D. Histone deacetylases in viral infections. Clin. Epigenetics 2020, 1–2, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Schnell, A.P.; Kohrt, S.; Thoma-Kress, A.K. Latency Reversing Agents: Kick and Kill of HTLV-1? Int. J. Mol. Sci. 2021, 22, 5545. [Google Scholar] [CrossRef]
- Lopes, J.R.; Chiba, D.E.; Dos Santos, J.L. HIV latency reversal agents: A potential path for functional cure? Eur. J. Med. Chem. 2021, 213, 113213. [Google Scholar] [CrossRef]
- Yang, H.; Wallace, Z.; Dorrell, L. Therapeutic Targeting of HIV Reservoirs: How to Give T Cells a New Direction. Front. Immunol. 2018, 9, 2861. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gonzales, S.; Colomer-Lluch, M.; Serra-Moreno, R. Barriers for HIV Cure: The Latent Reservoir. AIDS Res. Hum. Retrovir. 2018, 9, 739–759. [Google Scholar] [CrossRef]
- Abner, E.; Stoszko, M.; Zeng, L.; Chen, H.-C.; Izquierdo-Bouldstridge, A.; Konuma, T.; Zorita, E.; Fanunza, E.; Zhang, Q.; Mahmoudi, T.; et al. A new quinoline BRD4 inhibitor targets a distinct latent HIV-1 reservoir for reactivation from other “Shock” drugs. J. Virol. 2018, 92, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, G.; Mota, T.M.; Li, S.; Tumpach, C.; Lee, M.Y.; Jacobson, J.; Harty, L.; Anderson, J.L.; Lewin, S.R.; Purcell, D.F.J. HIV latency reversing agents act through Tat post translational modifications, Retrovirology. Retrovirology 2018, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 5, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Conery, A.R.; Sims, R.J. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 8, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Keck, K.M.; Moquin, S.A.; He, A.; Fernandez, S.G.; Somberg, J.J.; Liu, S.M.; Martinez, D.M.; Miranda, J.L. Bromodomain and extraterminal inhibitors block the Epstein-Barr virus lytic cycle at two distinct steps. J. Biol. Chem. 2017, 32, 13284–13295. [Google Scholar] [CrossRef] [Green Version]
- Gilham, D.; Smith, A.L.; Fu, L.; Moore, D.; Muralidharan, A.; Reid, S.; Stotz, S.; Johansson, J.; Sweeney, M.; Wong, N.; et al. Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-Cov-2 Infection In Vitro. Biomedicines 2021, 9, 437. [Google Scholar] [CrossRef]
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 2013, 87, 3871–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Li, Q.; Lievens, S.; Tavernier, J.; You, J. Abrogation of the Brd4-positive transcription elongation factor B complex by papillomavirus E2 protein contributes to viral oncogene repression. J. Virol. 2010, 84, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef]
- Rice, A.P. The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr. Pharm. Des. 2017, 23, 4098–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaware, N.; Zhou, M.M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Hewings, D.S.; Wang, M.; Philpott, M.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Vuppusetty, C.; Marsden, B.; Knapp, S.; et al. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54, 6761–6770. [Google Scholar] [CrossRef]
- Ren, Q.; Gao, W. Current status in the discovery of dual BET/HDAC inhibitors. Bioorg. Med. Chem. Lett. 2021, 31, 127671. [Google Scholar] [CrossRef] [PubMed]
- Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers 2019, 11, 304. [Google Scholar] [CrossRef] [Green Version]
- Shao, M.; He, L.; Zheng, L.; Huang, L.; Zhou, Y.; Wang, T.; Chen, Y.; Shen, M.; Wang, F.; Yang, Z.; et al. Structure-based design, synthesis and in vitro antiproliferative effects studies of novel dual BRD4/HDAC inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 4051–4055. [Google Scholar] [CrossRef]
- Zhang, Z.; Hou, S.; Chen, H.; Ran, T.; Jiang, F.; Bian, Y.; Zhang, D.; Zhi, Y.; Wang, L.; Zhang, L.; et al. Targeting epigenetic readerand eraser: Rational design, synthesis and in vitro evaluation of dimethyli-soxazoles derivatives as BRD4/HDAC dual inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2931–2935. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- McKeown, M.R.; Shaw, D.L.; Fu, H.; Liu, S.; Xu, X.; Marineau, J.J.; Huang, Y.; Zhang, X.; Buckley, D.L.; Kadam, A.; et al. Biased multicomponent reactions to develop novel bromodomain inhibitors. J. Med. Chem. 2014, 57, 9019–9027. [Google Scholar] [CrossRef] [Green Version]
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Lai, W.-H.; Zhu, L.; Li, W.; Wei, L.; Lee, K.-H.; Xie, L.; Chen, C.-H. Elimination of HIV-1 Latently Infected Cells by Gnidimacrin and a Selective HDAC Inhibitor. ACS Med. Chem. Lett. 2018, 9, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 3, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 6–7, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Clementino, L.D.C.; Fernandes, G.F.S.; Prokopczyk, I.M.; Laurindo, W.C.; Toyama, D.; Motta, B.P.; Baviera, A.M.; Henrique-Silva, F.; dos Santos, J.L.; Graminha, M.A.S. Design, synthesis and biological evaluation of N-oxide derivatives with potent in vivo antileishmanial activity. PLoS ONE 2021, 16, e0259008. [Google Scholar] [CrossRef]
- Gediya, L.K.; Belosay, A.; Khandelwal, A.; Purushottamachar, P.; Njar, V.C.O. Improved synthesis of histone deacetylase inhibitors (HDIs) (MS-275 and CI-994) and inhibitory effects of HDIs alone or in combination with RAMBAs or retinoids on growth of human LNCaP prostate cancer cells and tumor xenografts. Bioorg. Med. Chem. 2008, 6, 3352–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabal, O.; Sánchez-Arias, J.A.; Cuadrado-Tejedor, M.; De Miguel, I.; Pérez-González, M.; García-Barroso, C.; Ugarte, A.; De Mendoza, A.E.-H.; Sáez, E.; Espelosin, M.; et al. Discovery of in Vivo Chemical Probes for Treating Alzheimer’s Disease: Dual Phosphodiesterase 5 (PDE5) and Class I Histone Deacetylase Selective Inhibitors. ACS Chem. Neurosci. 2019, 10, 1765–1782. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Jiang, Y.; Wu, J.; Inks, E.S.; Chou, C.J.; Gao, S.; Hou, J.; Ding, Q.; Li, J.; et al. Selective HDAC inhibitors with potent oral activity against leukemia and colorectal cancer: Design, structure-activity relationship and anti-tumor activity study. Eur. J. Med. Chem. 2017, 134, 185–206. [Google Scholar] [CrossRef]
- Tian, Y.; Xie, Z.; Liao, C. Design, synthesis and anticancer activities of novel dual poly(ADP-ribose) polymerase-1/histone deacetylase-1 inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127036. [Google Scholar] [CrossRef]
- Wagner, F.F.; Lundh, M.; Kaya, T.; McCarren, P.; Zhang, Y.-L.; Chattopadhyay, S.; Gale, J.P.; Galbo, T.; Fisher, S.L.; Meier, B.C.; et al. An Isochemogenic Set of Inhibitors to Define the Therapeutic Potential of Histone Deacetylases in β-Cell Protection. ACS Chem. Biol. 2016, 11, 363–374. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Docking Score | ||
|---|---|---|---|
| HDAC-2 | HDAC-3 | BRD4 | |
| (1) | −8.451 | −8.260 | −2.956 |
| (2) | −10.238 | −8.490 | −5.731 |
| (3) | −6.940 | −8.209 | −4.839 |
| (4) | −9.001 | −8.608 | −6.014 |
| (5) | −9.003 | −4.895 | |
| (6) | −10.101 | −5.338 | |
| (7) | −10.036 | −4.298 | |
| (8) | −11.101 | −4.021 | |
| (9) | −8.567 | −4.975 | |
| (10) | −8.459 | −7.548 | −4.320 |
| Compounds | Inhibitory Activity (%) | |||||
|---|---|---|---|---|---|---|
| HDAC-1 | HDAC-2 | HDAC-3 | HDAC-4 | HDAC-11 | BRD4 | |
| (1) | 89 | 82 | 77 | - | - | 0 |
| (2) | 95 | 91 | 18 | <5 | <5 | 14 |
| (3) | 8 | 10 | <5 | <5 | <5 | 25 |
| (4) | 75 | 72 | 57 | <5 | <5 | 21 |
| (5) | 11 | 10 | 23 | <5 | <5 | 20 |
| (6) | 95 | 88 | 16 | <5 | <5 | 17 |
| (7) | 8 | 10 | <5 | <5 | <5 | 25 |
| (8) | 92 | 76 | 7 | <5 | <5 | 12 |
| (9) | 9 | <5 | <5 | <5 | <5 | 9 |
| (10) | 15 | 8 | <5 | <5 | <5 | 5 |
| Vorinostat 1 | 90 | 89 | 90 | - | - | 0 |
| JQ-1 2 | - | - | - | - | - | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, J.R.; Prokopczyk, I.M.; Gerlack, M.; Man Chin, C.; Santos, J.L.D. Design and Synthesis of Hybrid Compounds as Epigenetic Modifiers. Pharmaceuticals 2021, 14, 1308. https://doi.org/10.3390/ph14121308
Lopes JR, Prokopczyk IM, Gerlack M, Man Chin C, Santos JLD. Design and Synthesis of Hybrid Compounds as Epigenetic Modifiers. Pharmaceuticals. 2021; 14(12):1308. https://doi.org/10.3390/ph14121308
Chicago/Turabian StyleLopes, Juliana Romano, Igor Muccilo Prokopczyk, Max Gerlack, Chung Man Chin, and Jean Leandro Dos Santos. 2021. "Design and Synthesis of Hybrid Compounds as Epigenetic Modifiers" Pharmaceuticals 14, no. 12: 1308. https://doi.org/10.3390/ph14121308
APA StyleLopes, J. R., Prokopczyk, I. M., Gerlack, M., Man Chin, C., & Santos, J. L. D. (2021). Design and Synthesis of Hybrid Compounds as Epigenetic Modifiers. Pharmaceuticals, 14(12), 1308. https://doi.org/10.3390/ph14121308