Formation of New Alkynyl(phenyl)iodonium Salts and Their Use in the Synthesis of Phenylsulfonyl Indenes and Acetylenes

Abstract
:Introduction

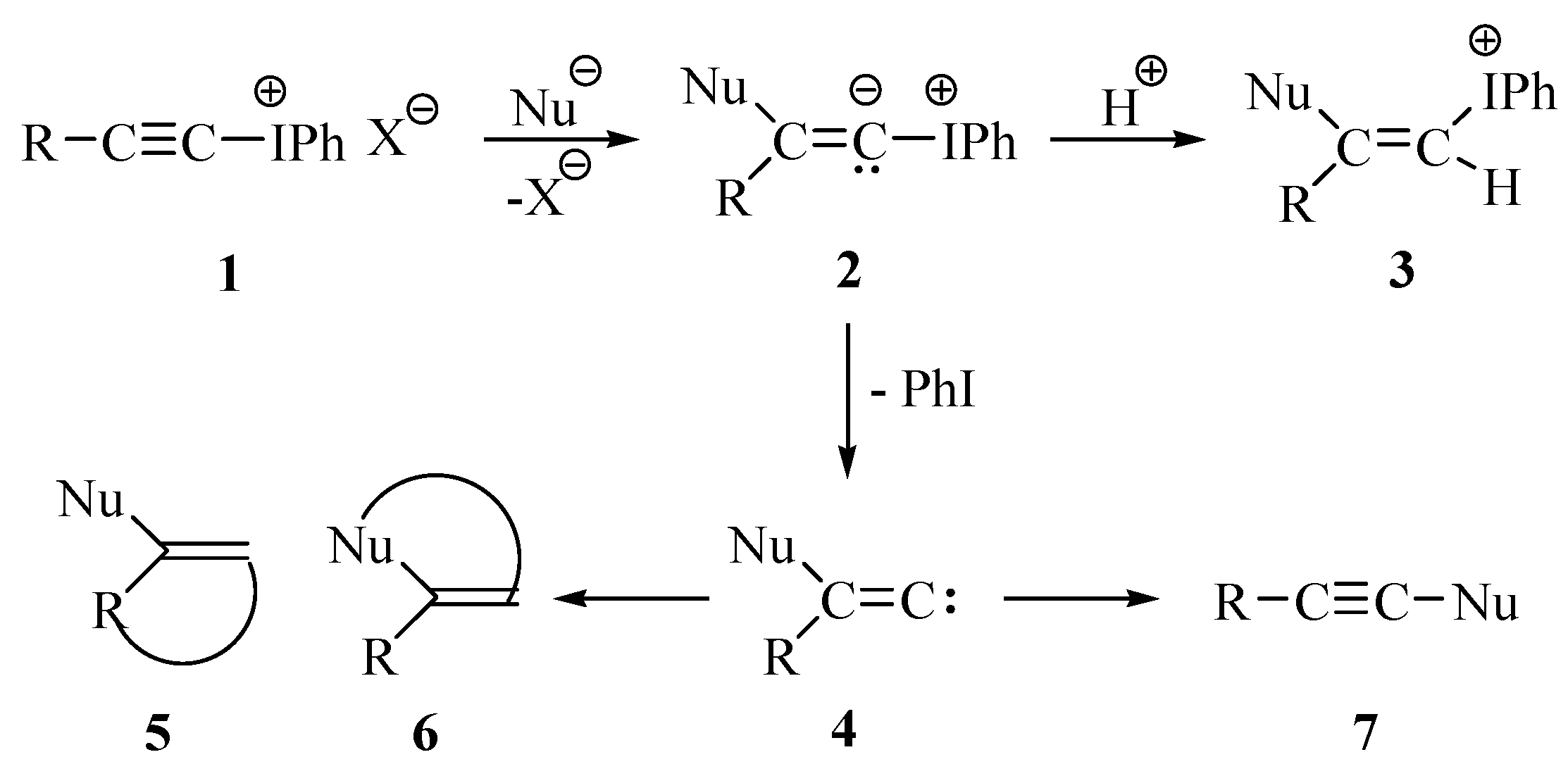
Results and Discussion





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
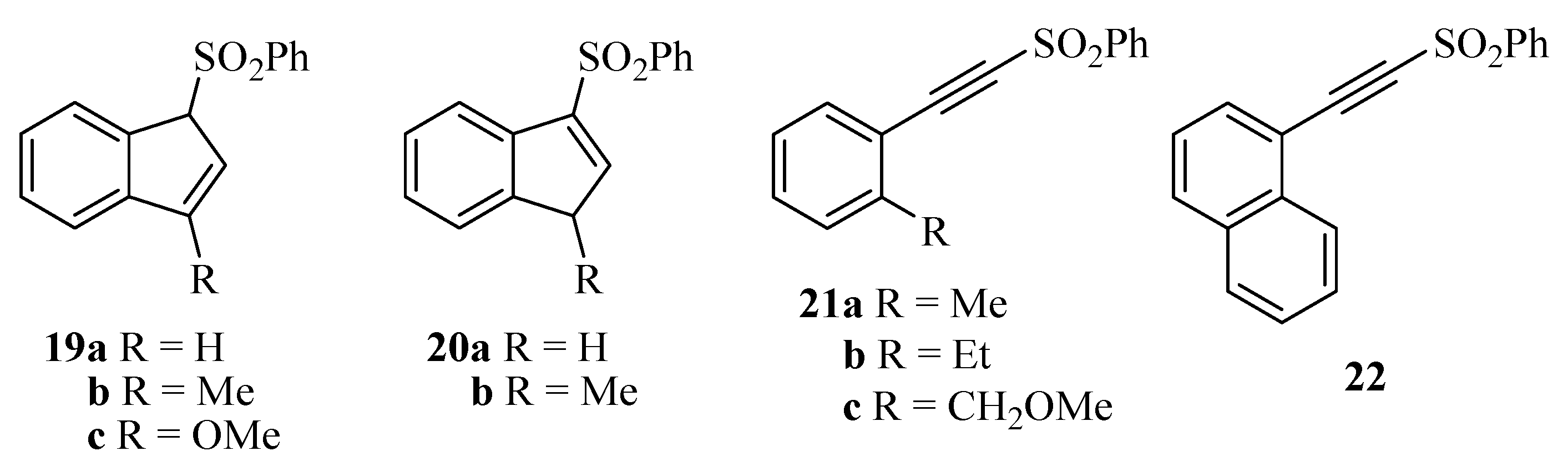
| Entry | Substrate | Ring type products ( A) | Rearrangement type products ( B) |
| 1 | 11a | 19a + 20a (32%, 5:1)a | 21a (26%) |
| 2 | 13a | 19a + 20a (44%, 5:1) | 21a (26%) |
| 3 | 11b | 19b (40%) | 21b (26%) |
| 4 | 13b | 19b (51%) | 21b (20%) |
| 5 | 11c | - | - |
| 6 | 13c | - | - |
| 7 | 17 | - | 22 (15%) |
| 8 | 18 | - | 22 (16%) |

Conclusions
Experimental
General
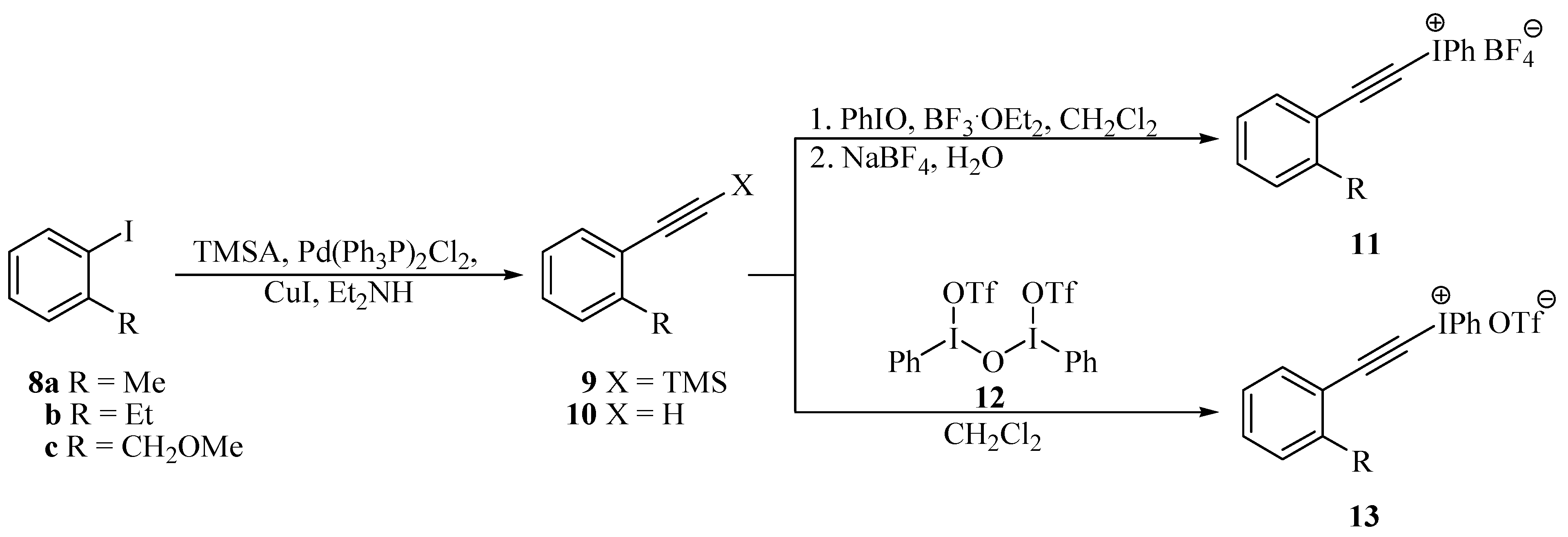
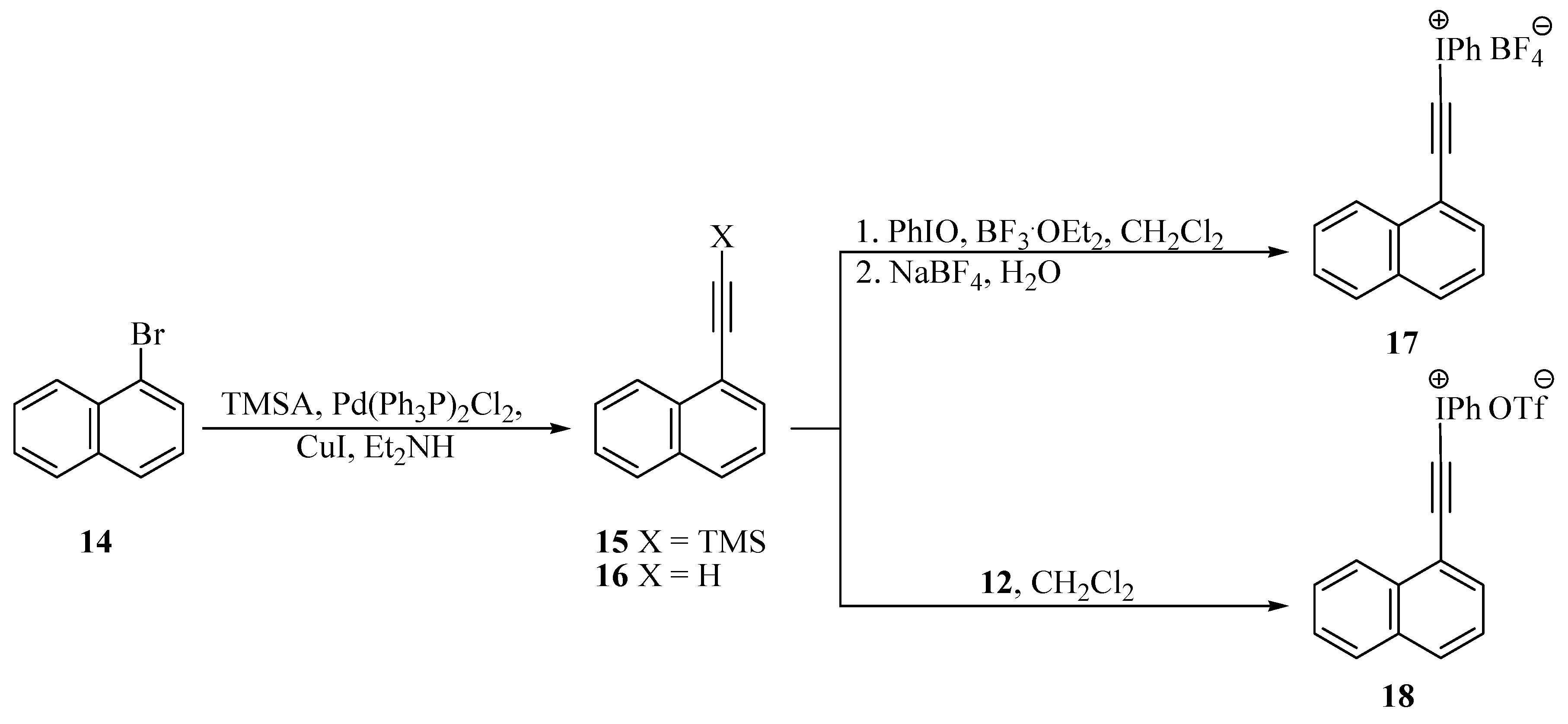
General procedure for the preparation of 2-aryl-trimethylsilylacetylenes 9 and 15 [23,24]
General procedure for the preparation of tetrafluoroborates 11 and 17 [26,27]
General procedure for the preparation of triflates 13 and 18 [29,30]
General procedure for the nucleophilic addition of sodium benzenesulfinate to iodonium salts
Acknowledgements
References and Notes
- Varvoglis, A. The Chemistry of Polycoordinated Iodine; VCH: New York, 1992; pp. 267–276. [Google Scholar]
- Varvoglis, A. Chemical Transformations Induced by Hypervalent Iodine Reagents. Tetrahedron 1997, 53, 1179–1255. [Google Scholar] [CrossRef]
- Kirmse, W. Alkenylidenes in Organic Synthesis. Angew. Chem. Int. Ed. 1997, 36, 1164–1170. [Google Scholar] [CrossRef]
- Moriarty, R.M.; Vaid, R.K. Carbon-Carbon Bond Formation Via Hypervalent Iodine Oxidations. Synthesis 1990, 431, 431–447. [Google Scholar] [CrossRef]
- Stang, P.J. Alkynyl- and Alkenyl(phenyl)iodonium Compounds. Angew. Chem. Int. Ed. 1992, 31, 274–285. [Google Scholar] [CrossRef]
- Stang, P.J.; Zdhankin, V.V. Organic Polyvalent Iodine Compounds. Chem. Rev. 1996, 96, 1123–1178. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, M.; Zdhankin, V.V. Topics in Current Chemistry: Hypervalent Iodine Chemistry; Wirth, T., Ed.; Springer: Berlin, 2003; pp. 52-57 and 120-130. [Google Scholar]
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, 1997; pp. 167–174. [Google Scholar]
- Zdhankin, V.V.; Stang, P.J. Alkynyliodonium Salts in Organic Synthesis. Tetrahedron 1998, 54, 10927–10966. [Google Scholar] [CrossRef]
- Marshall Beringer, F.; Galton, S.A. Acetylenic and Ethylenic Iodonium Salts and Their Reactions with a Carbanion. J. Org. Chem. 1965, 30, 1930–1934. [Google Scholar] [CrossRef]
- Koser, G.F.; Rebrovic, L.; Wettach, R.H. Functionalization of Alkenes and Alkynes with [Hydroxy(tosyloxy)iodo]benzene. Bis(tosyloxy)alkanes, Vinylaryliodonium Tosylates, and Alkynylaryliodonium Tosylates. J. Org. Chem. 1981, 46, 4324–4326. [Google Scholar] [CrossRef]
- Feldman, K.S.; Perkins, A.L. 1,6-C-H Insertion of Alkylidenecarbenes in 1-Naphthol and 1-Anthrol Derivatives. Tetrahedron Lett. 2001, 42, 6031–6033. [Google Scholar] [CrossRef]
- For a representative example, see: Tykwinski, R.R.; Whiteford, J.A.; Stang, P.J. Alkylidenecarbene Insertions into Aromatic C-H Bonds in Solution. J. Chem. Soc. Chem. Commun. 1993, 1800–1801. [Google Scholar]
- For a representative example, see: Williamson, B.L.; Tykwinski, R.R.; Stang, P.J. A New Method for the Synthesis of Cyclopentenones via the Tandem Michael Addition-Carbene Insertion Reaction of β-Ketoethynyl(phenyl)iodonium Salts. J. Am. Chem. Soc. 1994, 116, 93–98. [Google Scholar]
- For a representative example, see: Feldman, K.S.; Mareska, D.A. Alkynyliodonium Salts in Organic Synthesis. Preparation of Annelated Dihydropyrroles by Cascade Addition/Bicyclization of Dienyltosylamide Anions with Phenyl(propynyl)iodonium Triflate. J. Org. Chem. 1999, 64, 5650–5660. [Google Scholar]
- For a representative example, see: Feldman, K.S.; Bruendl, M.M.; Schildknegt, K. Preparation of Five-Membered Nitrogen-Containing Heterocycles via [Three-Atom + Two-Atom] Combination of Tosylamide Anions with Phenyl(propyny1)iodonium Triflate. J. Org. Chem. 1995, 60, 7722–7723. [Google Scholar]
- For a representative example, see: Wipf, P.; Venkatraman, S. A New Thiazole Synthesis by Cyclocondensation of Thioamides and Alkynyl(Aryl)Iodonium Reagents. J. Org. Chem. 1996, 61, 8004–8005. [Google Scholar]
- For a representative example, see: Nikas, S.; Rodios, N.; Varvoglis, A. The Reaction of Trimethylsilylethynyl(phenyl)iodonium Triflate with Some Phenolates: Formation of Substitution and sp2 C-H Insertion Products. Molecules 2000, 5, 1182–1186. [Google Scholar]
- Ochiai, M.; Kunishima, M.; Tani, S.; Nagao, Y. Generation of [β-(Phenylsulfony1)alkyli-dene]carbenes from Hypervalent Alkenyl- and Alkynyliodonium Tetrafluoroborates and Synthesis of 1 -(Phenylsulfony1)cyclopentenes. J. Am. Chem. Soc. 1991, 113, 3135–3142. [Google Scholar] [CrossRef]
- Tykwinski, R.R.; Stang, P.J.; Perksy, N.E. Preparation of Bis-Cyclopentene Ring Systems via Reaction of Bis[phenyl(iodonium)] Diyne Triflates with Soft Nucleophiles. Tetrahedron Lett. 1994, 35, 23–26. [Google Scholar] [CrossRef]
- Iodides 8a and 8b are commercially available.
- Iodide 8c was prepared according to: Larock, R.C.; Harisson, L.W.; Varvoglis, A. Mercury in Organic Chemistry. 26. Synthesis of Heterocycles via Intramolecular Solvomercuration of Aryl Acetylenes. J. Am. Chem. Soc. 1984, 106, 4218–4227. [Google Scholar]
- Takahashi, S.; Kuroyama, Y.; Sonogashira, K.; Hagihara, N. A Convenient Synthesis of Ethynylarenes and Di-ethynylarenes. Synthesis 1980, 627–631. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes, and Bromopyridines. Tetrahedron Lett. 1975, 4467–4470. [Google Scholar] [CrossRef]
- Saltzmann, H.; Sharefkin, J.G. Iodosobenzene. Organic Syntheses 1973, Coll. Vol. V, 658–659. [Google Scholar]
- Ochiai, M.; Kunishima, M.; Sumi, K.; Nagao, Y.; Fujita, E.; Arimoto, M.; Yamaguchi, H. Reaction of Alkynyltrimethylsilanes with a Hypervalent Organoiodine Compound: A New General Synthesis of Alkynyliodonium Salts. Tetrahedron Lett. 1985, 26, 4501–4504. [Google Scholar] [CrossRef]
- Ochiai, M.; Ito, T.; Takaoka, Y.; Masaki, Y.; Kunishima, M.; Tani, S.; Nagao, Y. Synthesis of Ethynyl(phenyl)iodonium Tetrafluoroborate. A New Reagent for Ethynylation of 1,3-Dicarbonyl Compounds. J. Chem. Soc. Chem. Commun. 1990, 118–119. [Google Scholar] [CrossRef]
- Hembre, R.T.; Scott, C.P.; Norton, J.R. Conversion of Olefins to Ditriflates by µ-Oxobis[(trifluoromethanesulfonato) (phenyl)iodine]. J. Org. Chem. 1987, 52, 3650–3654. [Google Scholar] [CrossRef]
- Bachi, M.D.; Bar-Ner, N.; Crittell, C.M.; Stang, P.J.; Williamson, B.L. Synthesis of Alkynyl(phenyl)iodonium Triflates and Their Reaction with Diethyl-2-Aminomalonate. J. Org. Chem. 1991, 58, 3912–3915. [Google Scholar] [CrossRef]
- Stang, P.J.; Arif, A.M.; Crittell, C.M. Ethynyl(phenyl)iodonium Triflate, [HC≡CIPh][OSO2CF3]: Preparation, Spectral Properties, Mechanism of Formation and X-ray Molecular Structure. Angew. Chem. Int. Ed. 1990, 29, 287–288. [Google Scholar] [CrossRef]
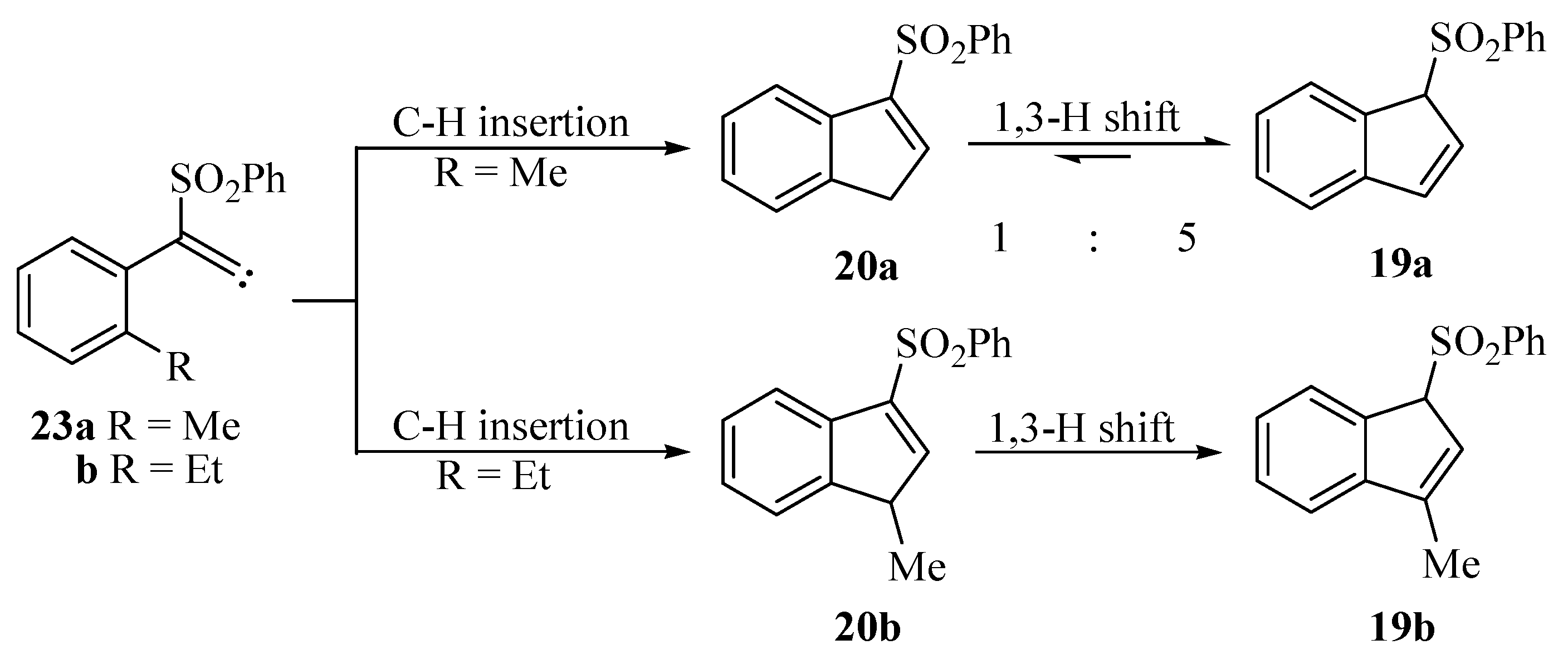
- The structures of 19a and 20a were proved unambiguously from the 1H-NMR spectra of their mixture. Thus, 19a has a peak at δ 5.07 which integrates for one H, whereas 20a gives a peak at δ 3.61 which integrates for two H. Similar conclusions were reached from their 13C-NMR spectra.
- 1H-NMR of the crude trifluroborate or triflate revealed that these salts were indeed formed.
- Feldman, K.S.; Wrobleski, M.L. Alkynyliodonium Salts in Organic Synthesis. Preparation of 2-Substituted-3-p-toluenesulfonyldihydrofurans from 1-Hydroxybut-3-ynyliodonium Ethers via a Formal Stevens Shift of a Carbon Group. Org. Lett. 2000, 2, 2603–2605. [Google Scholar] [CrossRef] [PubMed]
- See refs. [13], [16] and [18].
- Kitamura, T.; Zheng, L.; Taniguchi, H.; Sakurai, M.; Tanaka, R. Novel Cyclization to Benzofurans in the Reaction of Alkynyl(p-phenylene)bisiodonium Ditriflates with Phenoxide Anion. Tetrahedron Lett. 1993, 34, 4055–4058. [Google Scholar] [CrossRef]
- Kitamura, T.; Zheng, L.; Fukuoka, T.; Fujiwara, Y.; Taniguchi, H.; Sakurai, M.; Tanaka, R. Aromatic C-H Insertion of β-Phenoxyalkylidenecarbenes Generated by Reaction of Alkynyl(p-phenylene)bisiodonium Ditrifluoromethanosulfonates (Ditriflates) with Phenoxide Anions. J. Chem. Soc. Perkin Trans. 1997, 2, 1511–1515. [Google Scholar] [CrossRef]
- Kitamura, T.; Tsuda, K.; Fujiwara, Y. Novel Heteroaromatic C-H Insertion of Alkylidenecarbenes. A New Entry to Furopyridine Synthesis. Tetrahedron Lett. 1998, 39, 5375–5376. [Google Scholar] [CrossRef]
- D’Auria, M. Naturally Occurring 5-[(2-Thienyl)(ethynyl)]thiophene-2-carbaldehyde through a Short Synthesis of Diarylacetylenes. Synth. Commun. 1992, 22, 2393–2399. [Google Scholar] [CrossRef]
- Li, H.; Yang, H.; Petersen, J.L.; Wang, K.K. Biradicals/Zwitterions from Thermolysis of Enyne-Isocyanates. Application to the Synthesis of 2(1H)-Pyridones, Benzofuro[3,2-c]pyridin-1(2H)-ones, 2,5-Dihydro-1H-pyrido[4,3-b]indol-1-ones, and Related Compounds. J. Org. Chem. 2004, 69, 4500–4508. [Google Scholar] [CrossRef] [PubMed]
- Butler, I.R.; Soucy-Breau, C. Bipyridylacetylenes 1: The Synthesis of Some Bipyridylacetylenes via the Palladium-catalyzed Coupling of Acetylenes with 2,2´-Dibromobipyridyl, and the Single Crystal X-ray Structure of 6,6´-Biphenylethynyl-2,2´-bipyridine. Can. J. Chem. 1991, 69, 1117–1123. [Google Scholar] [CrossRef]
- Yoshiyuki, O.; Chellappa, K.L.; Kundu, S.K. Magnetic Shielding of Acetylenic Protons in Ethynylarenes. J. Org. Chem. 1972, 37, 3185–3187. [Google Scholar]
- Card, P.J.; Friendli, F.E.; Shecheter, H. Synthesis and Chemistry of 1H-Cyclo-buta[de]naphthalenes, 1-Alkylidene-1H-cyclobuta[de]naphthalenes, and 1H-Cyclobuta[de]na-phthalen-1-one. J. Am. Chem. Soc. 1983, 105, 6104–6114. [Google Scholar] [CrossRef]
- Sample availability: Contact the authors.
© 2005 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Koumbis, A.; Kyzas, C.; Savva, A.; Varvoglis, A. Formation of New Alkynyl(phenyl)iodonium Salts and Their Use in the Synthesis of Phenylsulfonyl Indenes and Acetylenes. Molecules 2005, 10, 1340-1350. https://doi.org/10.3390/10101340
Koumbis A, Kyzas C, Savva A, Varvoglis A. Formation of New Alkynyl(phenyl)iodonium Salts and Their Use in the Synthesis of Phenylsulfonyl Indenes and Acetylenes. Molecules. 2005; 10(10):1340-1350. https://doi.org/10.3390/10101340
Chicago/Turabian StyleKoumbis, A., C. Kyzas, A. Savva, and A. Varvoglis. 2005. "Formation of New Alkynyl(phenyl)iodonium Salts and Their Use in the Synthesis of Phenylsulfonyl Indenes and Acetylenes" Molecules 10, no. 10: 1340-1350. https://doi.org/10.3390/10101340
APA StyleKoumbis, A., Kyzas, C., Savva, A., & Varvoglis, A. (2005). Formation of New Alkynyl(phenyl)iodonium Salts and Their Use in the Synthesis of Phenylsulfonyl Indenes and Acetylenes. Molecules, 10(10), 1340-1350. https://doi.org/10.3390/10101340