Results and Discussion
2-Methylfuran (
2) is commercially available, whereas the dienophile, methyl-3-bromopropiolate (
3) had to be prepared by the reaction of methyl propiolate with
N-bromosuccinimide in acetone with silver nitrate as catalyst (86% yield) [
6]. The methyl propiolate was previously prepared by esterification of propiolic acid with methanol/sulfuric acid (2 days, 65% yield).
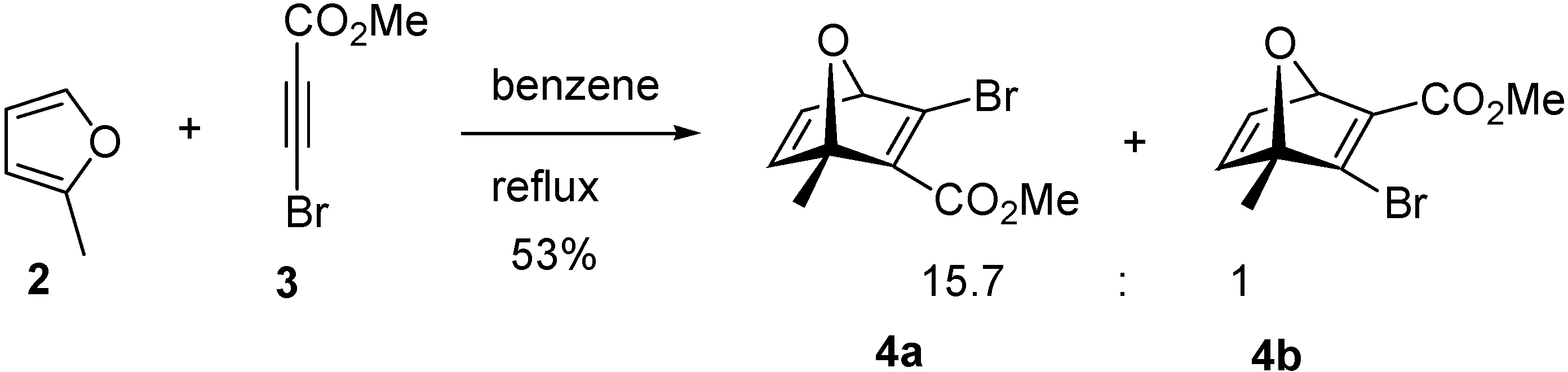
The Diels-Alder reaction between the diene
2 and dienophile
3 (benzene, 24 h reflux,
Scheme 2) resulted in a 15.7:1 mixture of the regioisomers
4a and
4b, respectively, which were separated by column chromatography and analyzed by nuclear magnetic resonance (NMR), with the assignment of all hydrogens, carbons and coupling constant values being accomplished using a combination of
1H-NMR,
13C-NMR, gsHMQC and gsHMBC. A remarkable aspect of this reaction is the high regioselectivity observed.
The treatment of adduct
4a with sodium methoxide in methanol at room temperature gave ketal
5a, the
endo product, as expected for a kinetic protonation process [
7]. Compound
5a was isolated by column chromatography in 87% yield.
The next step was the hydrolysis of ketal
5a. Leroy, in his paper concerning the synthesis of analogous compounds [
3], emphasized the difficulties of the ketal hydrolysis step. After some attempts he found that the H
+ form of the perfluorosulfonic acid resin Nafion
®-501, (Nafion-H), led to the corresponding ketone in 68-83% yield. However, this reagent is expensive and requires long reaction times (4 days).
We investigated several reagents and reaction conditions such as oxalic or sulfuric acid on wet silica gel, lithium tetrafluoroborate in wet acetonitrile, Amberlyst
®-15 in wet acetone, but none led to the desired hydrolysis. The treatment of
5a with PPTS (pyridinium
p-toluenesulfonate) gave less than 10% of
1, and required a large (tenfold) excess of PPTS and 16 hours at reflux [
8]. Finally, we found that in conc. HCl solution in methanol (room temperature, 7 hours), ketal
5a was converted to ketone
1 in 82% yield. The unusual lack of reactivity of
5a can be attributed to the retarding inductive effect of the 7-oxa bridge on the formation of cationic intermediates at C-3.
However, the double bond of
5a also seems to play a stabilizing effect on the intermediate carbocation (possibly through conjugation forming a non-classical ion, as shown in
Scheme 3), because after reduction of the double bond (H
2/Pd), the hydrolysis occurred readily providing a complex mixture from which only 7% of the corresponding ketone could be isolated (
Scheme 4).
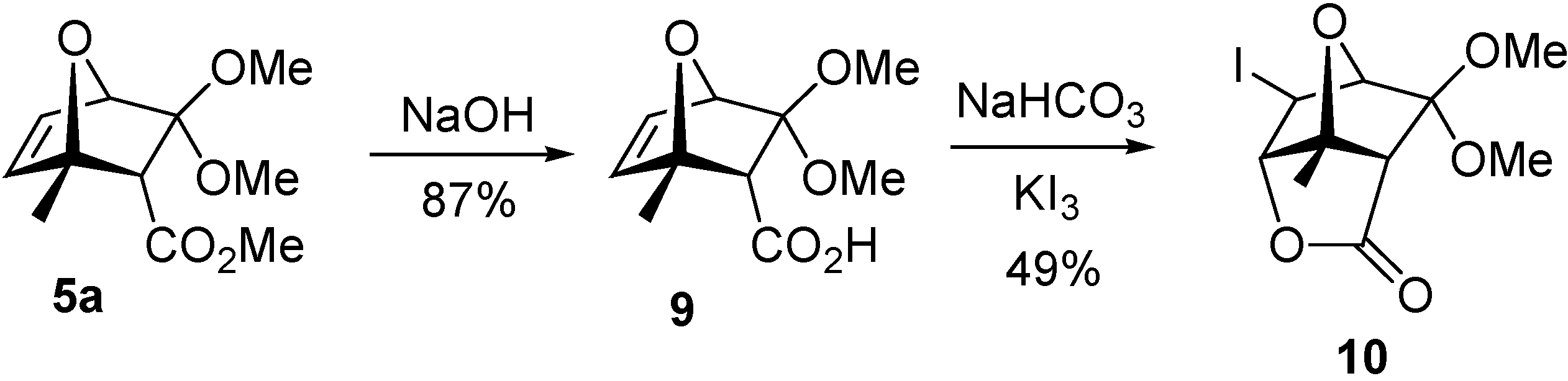
Further support for a favorable role of the double bond in the hydrolysis was provided by the fact that we found that lactone
10 [
9] (which was prepared from
5a as shown in
Scheme 5 to confirm its stereochemistry) did not undergo any transformation when treated with conc. HCl solution for 4 days.
Experimental
General
Melting points were determined on a Reichert Kofler block melting point apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were measured using a Bruker DPX-400 instrument (at 400 MHz and 100 MHz, respectively); deuteriochloroform was used as solvent and tetramethylsilane as an internal standard. IR spectra were measured with a Perkin-Elmer Spectrum RX IFTIR System. TLC was performed on precoated silica gel 60 F254 plates (0.25 mm thick, Merck), and silica gel 60 (70-230 mesh, Merck) was used for column chromatography.
(1S*,4R*)-3-Bromo-1-methyl-7-oxabicyclo[2.2.1]hepta-2,5-diene-2-carboxylic acid methyl ester (4a) and (1R*,4S*)-3-bromo-4-methyl-7-oxabicyclo[2.2.1]hepta-2,5-diene-2-carboxylic acid methyl ester (4b)
2-Methylfuran (2, 0.986 g, 12.02 mmol), methyl 3-bromopropiolate (3, 1.96 g, 12.02 mmol) and dry benzene (4 mL) were mixed and stirred at room temperature for 12 h. Another portion of 2-methyl-furan (2, 0.493 g, 6.01 mmol) was added and the resulting mixture was then heated at reflux for 12 h. The solvent was removed under vacuum and the residue was purified by chromatography on a silica gel column, eluting with hexane/ethyl acetate (9:1) to yield 1.46 g (50 %) of 4a and 0.093 g (3.2 %) of 4b. Compound 4a: 1H-NMR δ: 1.82 (s, 3H), 3.72 (s, 3H), 5.14 (d, 1H, J = 1.8 Hz), 6.92 (d, 1H, J = 5.3 Hz), 7.07 (dd, 1H, J1 = 5.3 Hz, J2 = 1.8 Hz); 13C-NMR δ: 16.8 (CH3), 52.0 (CH3), 88.4 (CH), 94.4 (C), 142.5 (CH), 144.8 (C), 147.4 (CH), 150.3 (C), 163.9 (C=O); IR (thin film) cm-1: 2933, 1722, 1607, 1442, 1306, 1260, 1099; MS m/z (relative intensity): 189 [M+ - 56] (7%), 82 (12%), 59 (24%), 51 (36%), 43 (100%), 15 (84%); Compound 4b: 1H-NMR δ: 1.74 (s, 3H), 3.79 (s, 3H), 5.62 (d, 1H, J = 2.0 Hz), 6.90 (d, 1H, J = 5.3 Hz), 7.20 (dd, 1H, J1 = 5.3 Hz, J2 = 2.0 Hz); 13C-NMR δ: 15.6 (CH3), 51.8 (CH3), 83.7 (CH), 95.1 (C), 144.1 (C), 144.5 (CH), 145.2 (CH), 153.2 (C), 162.9 (C=O).
(1S*,2R*,4R*)-3,3-Dimethoxy-1-methyl-7-oxabicyclo[2.2.1]hept-5-ene-2-carboxylic acid methyl ester (5a)
A solution of compound 4a (1.01 g, 4.12 mmol) in methanol (6 mL) was added dropwise to a 1M solution of sodium methoxide in methanol (20 mL), cooled with an ice bath. The reaction mixture was stirred for 2 h and then allowed to warm slowly to room temperature. The mixture was cooled again to 4 °C and then treated with 1:1 aqueous hydrochloric acid solution until pH 5. The methanol was removed under vacuum and the products were extracted with ethyl ether. The combined organic extracts were washed with saturated brine and dried over anhydrous MgSO4. The solvent was removed under vacuum and the residue was purified by chromatography in a silica gel column, eluting with hexane/ethyl acetate (7:3), yielding 0.818 g (87%) of 5a as a yellow solid; m.p. 60-62 °C; 1H-NMR δ: 1.54 (s, 3H), 2.90 (s, 1H), 3.14 (s, 3H), 3.40 (s, 3H), 3.63 (s, 3H), 4.66 (d, 1H, J = 1.8 Hz), 6.36 (dd, 1H, J1 = 5.8 Hz, J2 = 1.8 Hz), 6.61 (d, 1H, J = 5.8 Hz); 13C-NMR δ: 18.2 (CH3), 50.7 (CH3), 51.0 (CH3), 51.7 (CH3), 59.0 (CH), 84.0 (CH), 87.6 (C), 113.0 (C), 131.2 (CH), 140.8 (CH), 169.7 (C=O); IR (thin film) cm-1: 2953, 2837, 1738; 1436, 1316, 1142, 1060; MS m/z (relative intensity): 146 [M+ - 82] (12%), 115 (31%), 82 (8%), 69 (11%), 59 (9%), 28 (100%), 15 (22%).
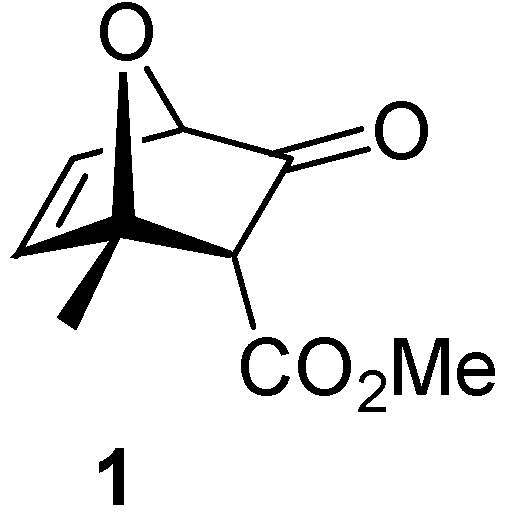
(1S*,2R*,4R*)-1-Methyl-3-oxo-7-oxabicyclo[2.2.1]hept-5-ene-2-carboxylic acid methyl ester (1)
Conc. hydrochloric acid solution (1.0 mL) was added dropwise at room temperature to a solution of compound 5a (103.5 mg; 0.454 mmol) in methanol (1.5 mL). The reaction mixture was stirred for 7 h. The resultant mixture was treated with water (1 mL) and extracted with ethyl ether. The organic extracts were washed with saturated NaHCO3 solution, then dried over anhydrous MgSO4 and the solvent was removed under vacuum, yielding 67.5 mg (82%) of a colorless oil; 1H-NMR δ: 1.74 (s, 3H), 3.15 (s, 1H), 3.74 (s, 3H), 4.76 (d, 1H, J = 2.0 Hz), 6.40 (dd, 1H, J1 = 5.6 Hz, J2 = 2.0 Hz), 6.75 (d, 1H, J = 5.6 Hz); 13C-NMR δ: 18.4 (CH3), 52.6 (CH3), 55.1 (CH), 83.5 (CH), 87.9 (C), 129.4 (CH), 144.3 (CH), 167.8 (C=O), 199.4 (C=O); IR (thin film) cm-1: 2956, 2840, 1732, 1436, 1303, 1205, 1023.
(1S*,2R*,4R*)-3,3-Dimethoxy-1-methyl-7-oxabicyclo[2.2.1]heptane-2-carboxylic acid methyl ester (6)
A solution of compound 5a (161 mg, 0.707 mmol) in methanol (1 mL) was placed in a 150 mL stainless steel pressure reactor. After addition of 5% Pd/C (25 mg), the resulting mixture was stirred under a hydrogen atmosphere at 6 atm and room temperature for 3 h. The reaction mixture was filtered through silica gel and the residue was washed with ethyl acetate. The organic extracts were dried over anhydrous MgSO4 and the solvent was removed under vacuum, yielding 126 mg (78%) of a colorless oil; 1H-NMR δ: 1.40 (ddt, 1H, J1 = J2 = 12.5 Hz, J3 = 5.0 Hz, J4 = 2.0 Hz), 1.48 (s, 3H), 1.77 (ddt, 1H, J1 = J2 = 12.5 Hz, J3 = 5.8 Hz, J4 = 4.5 Hz), 1.92 (ddd, 1H, J1 = 12.5 Hz, J2 = 9.6 Hz, J3 = 5.0 Hz), 2.80 (d, 1H, J = 2.0 Hz), 2.80 (ddd, 1H, J1 = 12.5 Hz, J2 = 9.6 Hz, J3 = 4.5 Hz), 3.19 (s, 3H), 3.33 (s, 3H), 3.70 (s, 3H), 4.37 (d, 1H, J = 5.8 Hz); 13C-NMR δ: 23.3 (CH3), 24.9 (CH2), 30.0 (CH2), 49.1 (CH3), 51.0 (CH3), 51.6 (CH3), 59.8 (CH), 83.1 (CH), 86.7 (C), 109.7 (C), 169.4 (C=O); IR (thin film) cm-1: 2951, 2837, 1734, 1213, 1151, 1071.
(1S*,2R*,4R*)-3,3-Dimethoxy-1-methyl-7-oxabicyclo[2.2.1]hept-5-ene-2-carboxylic acid (9)
Compound 5a (102.1 mg, 0.448 mmol) was stirred with 5% NaOH solution (2.6 mL) cooled with an ice bath for 4.5 h or until complete dissolution of 5a. The resultant mixture was diluted with water (8.5 mL) and washed with light petroleum (8.5 mL). The aqueous phase was cooled to 4 °C, treated with 1:1 aqueous HCl to pH 2 and then the product was extracted with chloroform. The organic extracts were dried over anhydrous MgSO4 and the solvent was removed under vacuum, yielding 82.8 mg (87%) of a white solid; m.p. 176-178 °C; 1H-NMR δ 1.65 (s, 3H), 2.96 (s, 1H), 3.27 (s, 3H), 3.44 (s, 3H), 4.74 (d, 1H, J = 1.8 Hz), 6.45 (dd, 1H, J1 = 5.8 Hz, J2 = 1.8 Hz), 6.57 (d, 1H, J = 5.8 Hz); 13C-NMR δ: 18.4 (CH3), 51.0 (CH3), 51.3 (CH3), 58.4 (CH), 84.0 (CH), 88.2 (C), 112.9 (C), 132.1 (CH), 140.7 (CH), 172.8 (C=O); IR (thin film) cm-1: 2937, 2825, 1700, 1559, 1278, 1143, 1054; MS m/z (relative intensity): 133 [M+ - 81] (37%), 87 (100%), 82 (75%), 69 (35%), 43 (49%).
(1S*,2S*,3S*,6R*,7R*)-2-Iodo-9,9-dimethoxy-7-mehyl-4,8-dioxatricyclo[4.2.1.03,7]nonan-5-one (10)
0.5 M aqueous NaHCO3 solution (0.75 mL) and a solution of I2 (63.5 mg, 0.25 mmol)/KI (124.5 mg, 0.75 mmol) in water (0.5 mL) were added to a solution of compound 9 (26.7 mg; 0.125 mmol) in THF (0.75 mL). The reaction mixture was protected from light and stirred at room temperature for 20 h. The mixture was extracted with ethyl ether, the organic extracts were washed with aqueous Na2S2O5 and saturated NaHCO3 solutions, then dried over anhydrous MgSO4 and the solvent was removed under vacuum, yielding 20.7 mg (49%) of a white solid; m.p. 199-201 °C; 1H-NMR δ: 1.65 (s, 3H), 2.96 (s, 1H), 3.25 (s, 3H), 3.28 (s, 3H), 4.22 (s, 1H), 4.52 (s, 1H), 4.73 (s, 1H); 13C-NMR δ: 17.1 (CH3), 21.8 (CH), 49.7 (CH3), 51.7 (CH3), 53.8 (CH), 87.5 (CH), 91.5 (CH), 91.9 (C), 109.5 (C), 171.0 (C=O); IR (thin film) cm-1: 2958, 2835, 1798, 1302, 1248, 1124, 1021.

{kind=link}
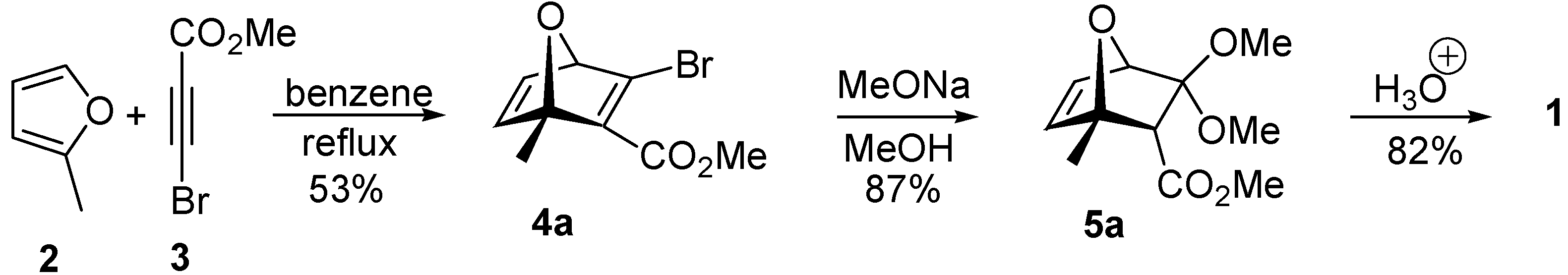{kind=link}
{kind=link}
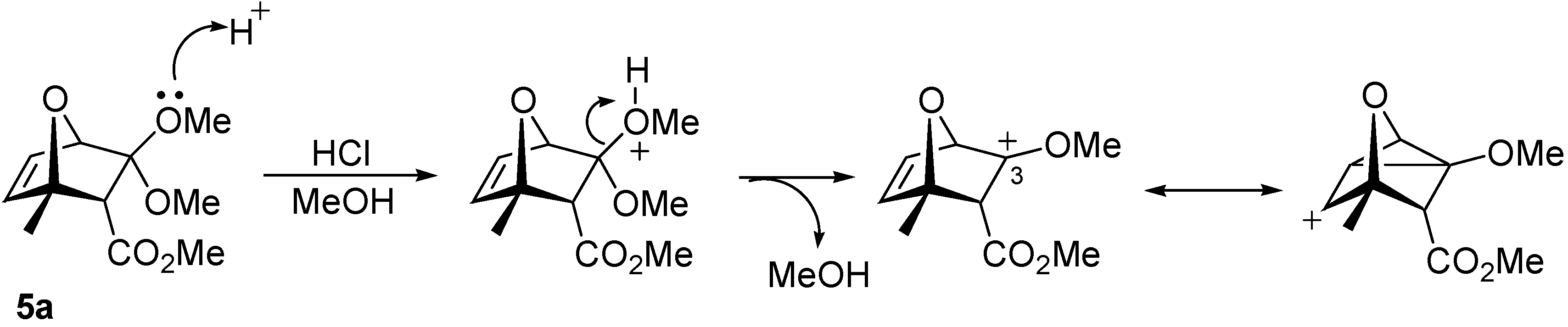{kind=link}
{kind=link}
{kind=link}





