Reactions of 9-Alkyl-3-aminocarbazoles with Ethyl-3-oxo-butanoate and Identification of the Products Obtained

Abstract
:Introduction
Results and Discussion


Conclusions
Experimental
General

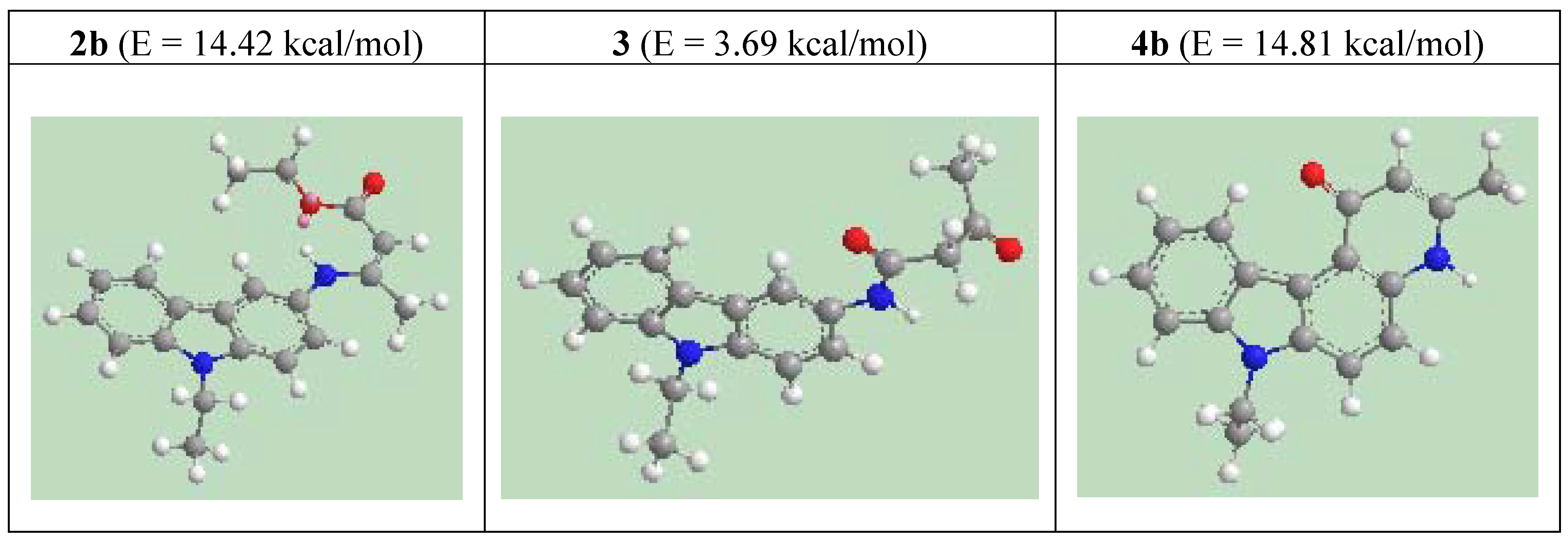{kind=link}
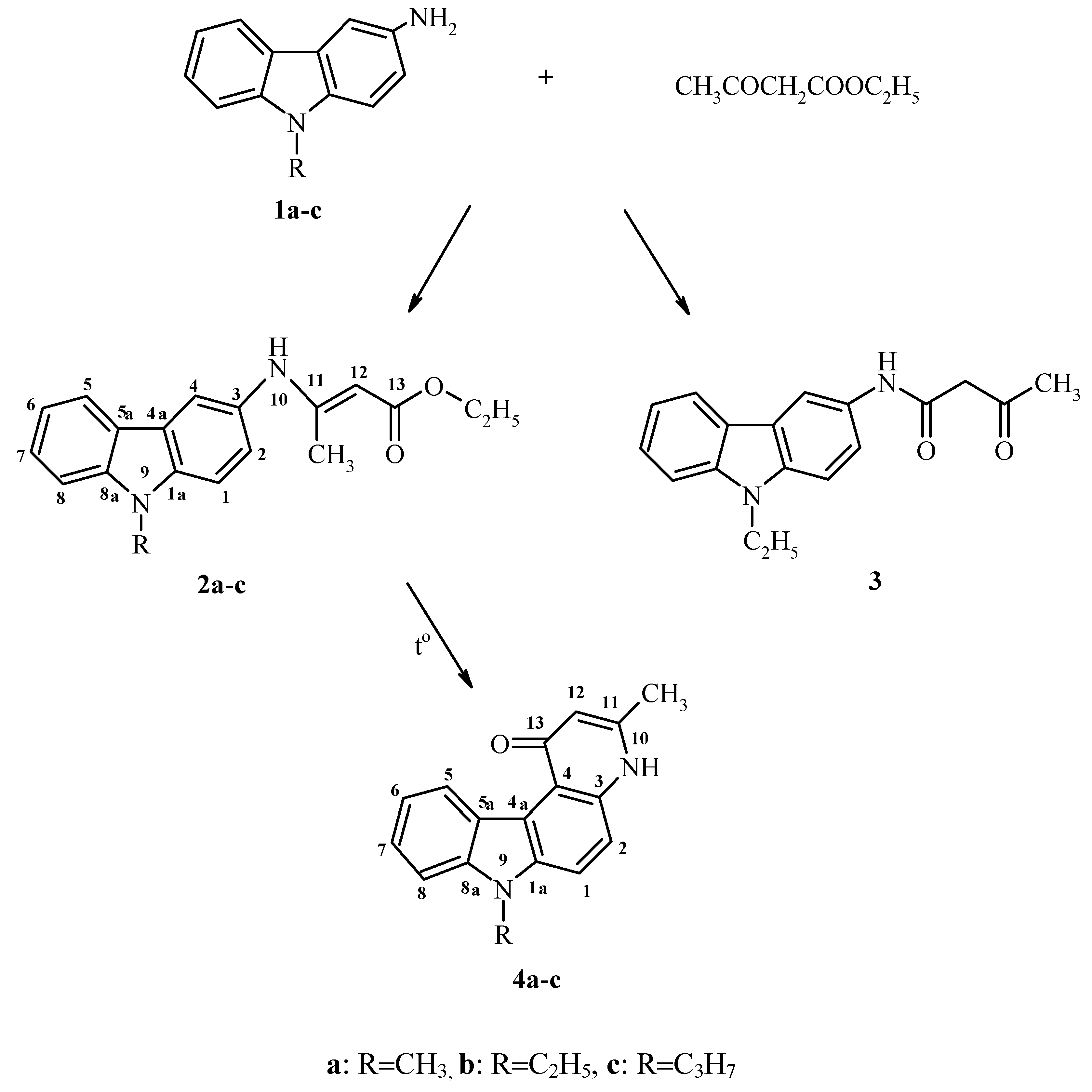{kind=link}
| Compound | 1H-NMR (solvent) | 13C-NMR (solvent) | IR (KBr tabl.) |
|---|---|---|---|
| 2a | (CD3)2CO): 1.24 (t, 3H, J3 = 7.1, COOCH2CH3), 1.97 (s, 3H, CCH3), 3.92 (s, 3H, NCH3), 4.11 (q, 2H, J = 7.1, COOCH2CH3), 4.67 (s, 1H, CH=), 7.21 (td, 1H, J3 = 6.8, J3 = 7.9, J4 = 1.3, Ar-6C-H), 7.30 (dd, 1H, J3 = 8.6, J4 = 2.0, Ar-2C-H), 7.48 (td, 1H, J3 = 6.8, J3 = 8.2, J4 = 1.3, Ar-7C-H), 7.53 (d, 1H, J3 = 8.6, Ar-1C-H), 7.54 (dd, IH, J3 = 8.2, J4 = 1.3, Ar-8CH), 7.98 (d, 1H, J4 = 2.0, Ar-4C-H), 8.17 (dd 1H, J3 = 7.9, J4 = 1.3, Ar-5C H), 10.48 (s, 1H, NH). | (CD3)2CO): 14.96 (COOCH2CH3), 20.18 (CCH3), 29.48 (NCH3), 58.80 (COOCH2CH3), 85.09 (CH=), 109.80 (Ar-C-8), 109.89 (Ar-C-1), 118.01 (Ar-C-4), 119.71 (Ar-C-6), 121.22 (Ar-C-5), 123.17 (Ar-C-5a), 123.78 (Ar-C-4a), 124.77 (Ar-C-2), 126.92 (Ar-C-7), 131.73 (Ar-C-3), 139.91 (Ar-C-1a), 142.52 (Ar-C-8a), 161.26 (C=), 170.94 (CO). | 3250(NH), 1656(CO). |
| 2b | (d6-DMSO): 1.22 (t, 3H, J = 7.1, COOCH2CH3), 1.32 (t, 3H, J = 7.1, NCH2CH3), 1.96 (s, 3H, CCH3), 4.08 (q, 2H, J = 7.1, COOCH2CH3), 4.43 (q, 2H, J = 7.1, NCH2CH3), 4.68 (s, 1H, CH=), 7.19 (td, 1H, J3 = 7.4, J3 = 7.6, J4= 1.3, Ar-6C-H), 7.29 (dd, 1H, J3 = 8.6, J4 = 2.0, Ar-2C-H), 7.46 (td, 1H, J3 = 6.6, J3 = 7.4, J4 = 1.3, Ar-7C-H), 7.58 (d, 1H, J3 = 8.6, Ar-1CH), 7.59 (dd, 1H, J3 = 6.6, J3 = 1.3, Ar-8C-H), 8.02 (d, 1H, J4 = 2.0, Ar-4C-H), 8.18 (dd, 1H, J3 = 7.6, J4 =1.3, Ar-5C-H), 10.38 (s, 1H, NH). | (d6-DMSO): 13.63 (NCH2CH3), 14.49 (COOCH2CH3), 19.76 (CCH3), 36.99 (NCH2CH3), 57.90 (COOCH2CH3), 83.93 (C=), 109.13 (Ar-C-8), 109.25 (Ar-C-1), 116.99 (Ar-C-4), 118.65 (Ar-C-6), 120.64 (Ar-C-5), 121.86 (Ar-C-5a), 122.42 (Ar-C-4a), 123.57 (Ar-C-2), 125.98 (Ar-C-7), 130.21 (Ar-C-3), 137.37 (Ar-C-1a), 140.03 (Ar-C-8a), 160.29 (C=), 169.48 (CO). | 3267(NH), 1652(CO). |
| 2c | (CDCl3): 0.98 (t, 3H, J=7.5, COOCH2CH3), 1.31 (t, 3H, J = 6.9, NCH2CH2CH3), 1.89 − 1.92 (m, 2H, NCH2CH2CH3), 1.94 (s, 3H, CCH3), 4.18 (q, 2H, J = 7.5, COOCH2CH3), 4.26 (q, 2H, J = 6.9, NCH2CH2CH3), 4.69 (s, 1H, CH=), 7.21 (dd, 1H, J3 = 8.2, J4 = 2.0, Ar-2C-H), 7.22 (td, 1H, J3 = 7.6, J3 = 8.2, J4 = 1.3, Ar-6C-H), 7.34 (dd, 1H, J3 = 6.9, J4 = 1.3, Ar-8C-H), 7.40 (d, 1H, J3 = 8.2, Ar-1C-H), 7.47 (td, 1H, J3 = 8.2, J3 = 6.9, J4 = 1.3, Ar-7C-H), 7.83 (d, 1H, J4 = 2.0, Ar-4C-H), 8.03 (dd, 1H, J3 = 7.6, J4 = 1.3, Ar-5C-H), 10.38 (s, 1H, NH). | (CDCl3): 11.82 (NCH2CH2CH3), 14.68 (COOCH2CH3), 20.29 (CCH3), 22.34 (NCH2CH2CH3), 44.78 NCH2CH2CH3), 58.60 (COOCH2CH3), 84.22 (C=), 108.79 (Ar-C-8), 108.93 (Ar-C-1), 117.79 (Ar-C-4), 118.93 (Ar-C-6), 120.39 (Ar-C-5), 122.37 (Ar-C-5a), 122.99 (Ar-C-4a), 124.26 (Ar-C-2), 126.04 (Ar-C-7), 130.70 (Ar-C-3), 138.58 (Ar-C-1a), 141.05 (Ar-C-8a), 160.76 (C=), 170.61 (CO). | 3257(NH), 1651(CO). |
| 3 | (CDCl3): 1.41 (t, 3H, J = 7.2, NCH2CH3), 2.36 (s, 3H, COCH3), 3.65 (s, 2H, COCH2), 4.32 (q, 2H, J = 7.2, NCH2CH3), 7.23 (td, 1H, J3 = 7.0, J3 = 7.8, J4 = 1.1, Ar-6C-H), 7.31 (d, 1H, J3 = 8.7, Ar-1C-H),7.40 (dd, 1H, J3 = 8.2, J4 = 1.1, Ar-8C-H), 7.49 (td, 1H, J3 = 8.2, J3 = 7.0, J4 = 1.1, Ar-7C-H), 7.55 (dd, 1H, J3 = 8.7, J4 = 2.0, Ar-2C-H), 8.08 (dd, 1H, J3 = 7.8, J4 = 1.1, Ar-5C-H), 8.36 (d, 1H, J4 = 2.0, Ar-4CH), 9.23 (s, 1H, NH). | (CDCl3): 14.06 (NCH2CH3), 31.45 (COCH3), 37.81 (NCH2CH3), 50.30 (COCH2CO), 108.71 (Ar-C-8), 108.79 (Ar-C-1), 113.26 (Ar-C-4), 119.01 or 119.86 (Ar-C-6), 119.01 or 119.86 (Ar-C-2), 120.96 (Ar-C-5), 122.99 (Ar-C-5a), 123.21 (Ar-C-4a), 126.12 (Ar-C-7), 129.60 (Ar-C-3), 137.55 (Ar-C-1a), 140.66 (Ar-C-8a), 163.93 (NHCO), 205.54 (COCH3). | 3276(NH), 1721(CO), 1640(CO). |
| 4a | (d6-DMSO): 2.41 (s, 3H, CCH3), 3.97 (s, 3H, NCH3), 6.09 (CH=), 7.18 (td, 1H, J3 = 7.1, J3 = 8.2, Ar-6CH), 7.46 (td, 1H, J3 = 8.2, J3 = 7.1, Ar-7C-H), 7.60 (d, 1H, J3 = 8.2, Ar-8C-H), 7.69 (d, 1H, J3 = 8.9, Ar-1CH), 8.02 (d, 1H, J3 = 8.9, Ar-2C-H), 9.94 (d, 1H, J3 = 8.2, Ar-5C-H), 11.69 (s, 1H, NH). | (d6-DMSO): 18.87 (CCH3), 29.01 (NCH3), 108.60 (CH=), 109.93 (Ar-C-1), 109.93 (Ar-C-8), 114.72 (Ar-C-2), 116.57 (Ar-C-4), 117.91 (Ar-C-6), 121.04 (Ar-C-4a), 121.04 (Ar-C-5a), 122.75 (Ar-C-5), 125.28 (Ar-C-7), 128.90 (Ar-C-3), 136.33 (Ar-C-1a), 140.70 (Ar-C-8a), 146.68 (C=), 177.97 (CO). | 3267(NH), 1623(CO). |
| 4b | (d6-DMSO): 1.28 (t, 3H, J = 7.2, NCH2CH3), 2.45 (s, 3H, CCH3), 4.52 (q, 2H, J = 7.2 NCH2CH3), 6.05 (s, 1H, CH=), 7.14 (t, 1H, J3 = 7.5, J3 = 8.2, Ar-6C-H), 7.42 (t, 1H, J3 = 7.5, J3 = 8.2, Ar-7C-H), 7.59 (d, 1H, J3 = 8.2, Ar-8C-H), 7.65 (d, 1H, J3 = 8,8, Ar-1C-H), 8.02 (d, 1H, J3 = 8.8, Ar-2C-H), 9.90 (d, 1H, J3 = 8.2, Ar-5C-H), 11.65 (s, 1H, NH). | (d6-DMSO): 13.91 (NCH2CH3), 18.89 (CH3), 36.86 (NCH2CH3), 108.52 (CH=), 109.95 (Ar-C-1), 109.95 (Ar-C-8), 114.67 (Ar-C-2), 116.62 (Ar-C-4), 117.93 (Ar-C-6), 121.19 (Ar-C-4a), 121.19 (Ar-C-5a), 122.93 (Ar-C-5), 125.29 (Ar-C-7), 129.10 (Ar-C-3), 136.34 (Ar-C-1a), 139.58 (Ar-C-8a), 146.58 (C=), 177.97 (CO). | 3281(NH), 1619(CO). |
| 4c | (d6-DMSO): 0.85 (t, J = 7.2, 3H, NCH2CH2CH3), 1.75 − 1.82 (m, 2H, NCH2CH2CH3), 2.37 (s, 3H, CCH3), 4.55 (t, J = 7.2, 2H, NCH2CH2CH3), 6.04 (s, 1H, CH=), 7.13 (t, 1H, J3 = 7.6, J3 = 8.2, Ar-6C-H), 7.41 (t, 1H, J3 = 7.6, J3 = 8.2, Ar-7CH), 7.60 (d, 1H, J3 = 8.2, Ar-8C-H), 7.64 (d, 1H, J3 = 8.8, Ar-1C-H), 8.03 (d, 1H, J3 = 8.8, Ar-2CH), 9.90 (d, 1H, J3 = 8.2, Ar-5C-H), 11.63 (s, 1H, NH). | (d6-DMSO): 11.31 (NCH2CH2CH3), 18.84 (CH3), 22.09 (NCH2CH2CH3), 43.53 (NCH2CH2CH3), 108.98 (CH=), 109.93 or 109.99 (Ar-C-1), 109.93 or 109.99 (Ar-C-8), 114.94 (Ar-C-2), 116.56 (Ar-C-4), 117.88 (Ar-C-6), 121.11 (Ar-C-5a), 121.11 (Ar-C-4a), 122.78 (Ar-C-5), 125.23 (Ar-C-7), 129.03 (Ar-C-3), 136.32 (Ar-C-1a), 140.13 (Ar-C-8a), 146.51 (C=), 177.98 (CO). | 3283(NH), 1643(CO). |
| Compound | m. p. (°C) | Molecular Formula | Elemental analysis data (Calculated / Found) % | Yield (%) | ||
|---|---|---|---|---|---|---|
| C | H | N | ||||
| 2a | 130 – 131 | C19H20N2O2 | 74.00 / 74.08 | 6.54 / 6.48 | 9.08 / 8.96 | 80.0 |
| 2b | 134 – 135 | C20H22N2O2 | 74.51 / 74.46 | 6.88 / 7.09 | 8.69 / 8.43 | 83.0 |
| 2c | 77 – 78 | C21H24N2O2 | 74.97 / 74.71 | 7.19 / 7.24 | 8.33 / 8.43 | 76.0 |
| 3 | 157 – 158 | C18H18N2O2 | 73.45 / 73.34 | 6.16 / 6.37 | 9.52 / 9.45 | 73.0 |
| 4a | > 330 | C17H14N2O | 77.84 / 77.63 | 5.38 / 5.10 | 10.68 / 10.45 | 63.0 |
| 4b | 254 – 256 | C18H16N2O | 78.24 / 78.20 | 5.84 / 5.82 | 10.14 / 10.09 | 46.0 |
| 4c | 274 – 275 | C19H18N2O | 79.90 / 79.86 | 4.59 / 4.62 | 9.81 / 9.92 | 69.0 |
References
- Suffness, M.; Cordell, G.A. The Alkaloids; Brossi, A., Ed.; Academic Press: New York, 2000; Vol. 54, p. 395. [Google Scholar]
- Saxton, J.E. Indoles; Wiley-Interscience: New York, 1983; Pt. 4; p. 201. [Google Scholar]
- Kleemann, A.; Engel, J. Pharmazeutische Wirkstoffe; Georg Thieme Verlag: Stuttgart-New York, 1978; Volume V, p. 555. [Google Scholar]
- Kutkevicius, S.; Stanisauskaite, A.; Getautis, V.; Railaite, A. Synthesis of Carbazole Containing Organic Photosemiconductors Using Dimercapto Compounds. J. Prakt. Chem. 1995, 337, 315–318. [Google Scholar]
- Zhang, Y.; Wada, T.; Wang, L.; Sasabe, H. A Novel Approach to the Synthesis of Conjugated Carbazole Trimers as Multifunctional Cromophores for Photorefractive Materials. Tetrahedron Lett. 1997, 38, 1785–1788. [Google Scholar] [CrossRef]
- Elderfield, R.C. Heterocyclic Compounds; Wiley: New York, 1952; Vol. 4, p. 430. [Google Scholar]
- Reynolds, G.A.; Hauser, C.R. Org. Synth. 1949, 29, 70–71.
- Hesse, M.; Meier, H.; Zeeh, B. Spectroscopic Methods in Organic Chemistry; Georg Thieme Verlag: Stuttgart-New York, 1997; pp. 103-124, 142-163, 174. [Google Scholar]
- Kalinowski, H.O.; Berger, S.; Braun, S. 13C-NMR-Spektroskopie; Georg Thieme Verlag: Stuttgart-New York, 1984; pp. 84-122, 134-149, 283-308, 359. [Google Scholar]
- Sapijanskaite, B.; Mickevicius, V.; Mikulskiene, G. Synthesis of 1-(9-alkyl-9H-carbazol-3-yl)-4-carboxy-2-pyrrolidinone Derivatives. Khim. Geterotsikl. Soedin. 2003, 9, 1305–1315. [Google Scholar]
- Claramunt, R.M.; Lopez, C.; Sanz, D.; Alkorta, I.; Elguero, J.A. Multinuclear NMR Spectral Study of Parent Azoles and Benzazoles: Experimental Results and GIAO AB INITIO Calculations. Heterocycles 2001, 55, 2109–2121. [Google Scholar] [CrossRef]
- Oliveira, M.M.; Carvalho, L.M.; Moustrou, C.; Samat, A.; Guglielmetti, R.; Oliveira-Campos, A.M.F. NMR Analysis of a Series of Photochromic Pyranocarbazoles. Magn. Reson. Chem. 2001, 39, 637–640. [Google Scholar] [CrossRef]
- Balsells, R.E. 13C NMR Spectra of Substituted Carbazoles and Azacarbazoles (β-Carbolines). Magn. Reson. Chem. 1983, 26, 1109–1112. [Google Scholar] [CrossRef]
- Borovik, V.P.; Shakirov, M.M.; Shkurko, O.P. 1H and 13C-NMR Spectra of 9H-pyrimido[4,5-b]indoles. Khim. Geterotsikl. Soedin. 2003, 10, 1531–1537. [Google Scholar]
- Giraud, J.; Marzin, C. Comparative 13C NMR study of some Deuteriated and Undeuteriated Molecules. Org. Magn. Reson. 1979, 12, 649–651. [Google Scholar]
- Welti, D.H. Revision of the 13C NMR Signal Assignment of Harman, Based on One- and Two-Dimensional INADEQUATE Spectroscopy. Magn. Reson. Chem. 1985, 23, 872–876. [Google Scholar] [CrossRef]
- Shamma, M.; Hindenlang, D.M. Carbon –13 NMR Shift Assignments of Amines and Alkaloids; Plenum Press: New York-London, 1979; p. 97. [Google Scholar]
- Chem 3D Ultra 9.0, Cambridge Scientific Software; Serial number: 031 406391 4800.
- Sample Availability: Available from the authors.
© 2006 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Sapijanskaite, B.; Mickevicius, Y.; Mikulskiene, G. Reactions of 9-Alkyl-3-aminocarbazoles with Ethyl-3-oxo-butanoate and Identification of the Products Obtained. Molecules 2006, 11, 72-80. https://doi.org/10.3390/11010072
Sapijanskaite B, Mickevicius Y, Mikulskiene G. Reactions of 9-Alkyl-3-aminocarbazoles with Ethyl-3-oxo-butanoate and Identification of the Products Obtained. Molecules. 2006; 11(1):72-80. https://doi.org/10.3390/11010072
Chicago/Turabian StyleSapijanskaite, Birute, Ytautas Mickevicius, and Gema Mikulskiene. 2006. "Reactions of 9-Alkyl-3-aminocarbazoles with Ethyl-3-oxo-butanoate and Identification of the Products Obtained" Molecules 11, no. 1: 72-80. https://doi.org/10.3390/11010072
APA StyleSapijanskaite, B., Mickevicius, Y., & Mikulskiene, G. (2006). Reactions of 9-Alkyl-3-aminocarbazoles with Ethyl-3-oxo-butanoate and Identification of the Products Obtained. Molecules, 11(1), 72-80. https://doi.org/10.3390/11010072