Conclusions
We have studied the feasibility and the conditions of the hybridization methodology for the synthesis of three new L-ascorbic ferulic acid hybrids. This methodology led us to introduce a chemical spacer connected to the hydroxyl in the C-3 position of the L-ascorbic acid and the phenol of the ferulic ester. The junction could be crucial for both stability and biological activity insofar as the enediol system is protected.
The thermal stability and the inhibitory effects of the products on tyrosinase activity, and on active oxygen species and free radicals in vitro, are currently under investigation and the results will be published elsewhere.
Experimental
General
Commercial reagents were purchased from Aldrich, Acros Organics and Alfa Aesar and used without additional purification. Melting points were determined on a Kofler heating bench and are uncorrected. IR spectra were recorded on a Perkin Elmer BX FT-IR spectrophotometer. Optical rotations were obtained on a Perkin-Elmer 343 polarimeter. 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were recorded in DMSO-d6 on a JEOL Lambda 400 spectrometer. Chemical shifts are expressed in parts per million downfield from tetramethylsilane as an internal standard and coupling constants in hertz. Mass spectra were taken on a JEOL JMS GCMate spectrometer at ionising potential of 70 eV (EI) or were performed using a spectrometer LC-MS Waters alliance 2695 (ESI+). Chromatography was carried out on a column using flash silica gel 60 Merck (0.063-0.200 mm) as the stationary phase. Thin-layer chromatography was performed on 0.2 mm precoated plates of silica gel 60F-264 (Merck) and spots were visualized using an ultraviolet-light lamp.
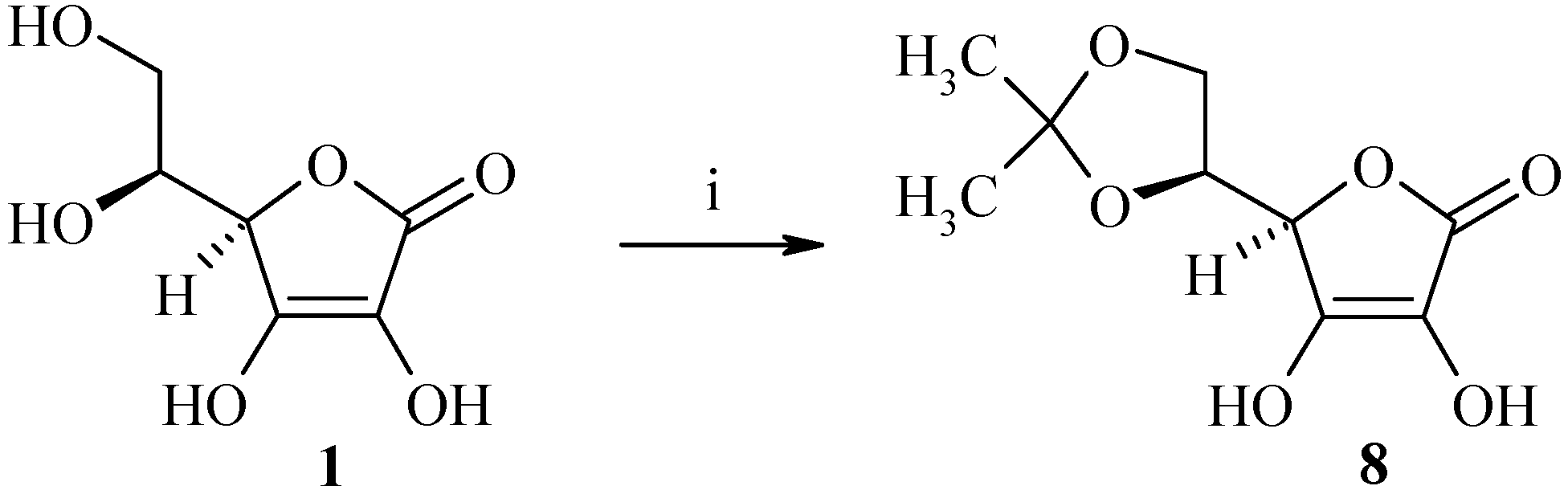
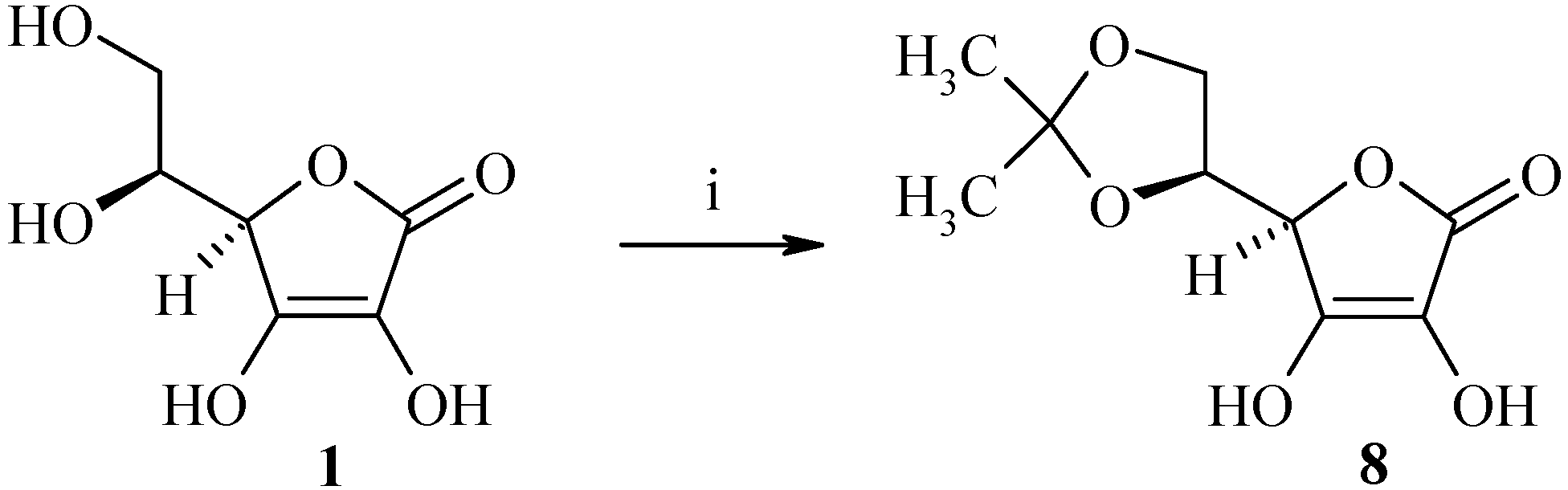
Synthesis of (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8)
Acetyl chloride (1 mL, 14.2 mmol) was added dropwise under nitrogen to a solution of L-ascorbic acid 1 (10 g, 56.8 mmol) in acetone (40 mL) and the solution was then stirred at room temperature for 3 h. The resulting white mixture was cooled to 4°C, filtered and then washed with acetone (2 x 20 mL) to afford 9.1 g of (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8) as a white solid (yield: 74%); m.p.: 210°C; IR (cm-1): 3242 (OH), 1754 (C=O), 1663 (C=C), 1333 (C(CH3)2), 1140 (dioxolane); 1H-NMR: 11.29 (bs, 1H, OH), 8.48 (bs, 1H, OH), 4.70-4.69 (m, 1H, H4), 4.25-4.23 (m, 1H, H5), 4.08-4.06 (m, 1H, H6), 3.87-3.86 (m, 1H, H6), 1.24 (s, 6H, CH3); 13C-NMR: 173.4, 157.1, 117.8, 110.4, 76.6, 76.4, 65.1, 26.3, 25.7; MS (ESI+) m/z 219 (M++1).
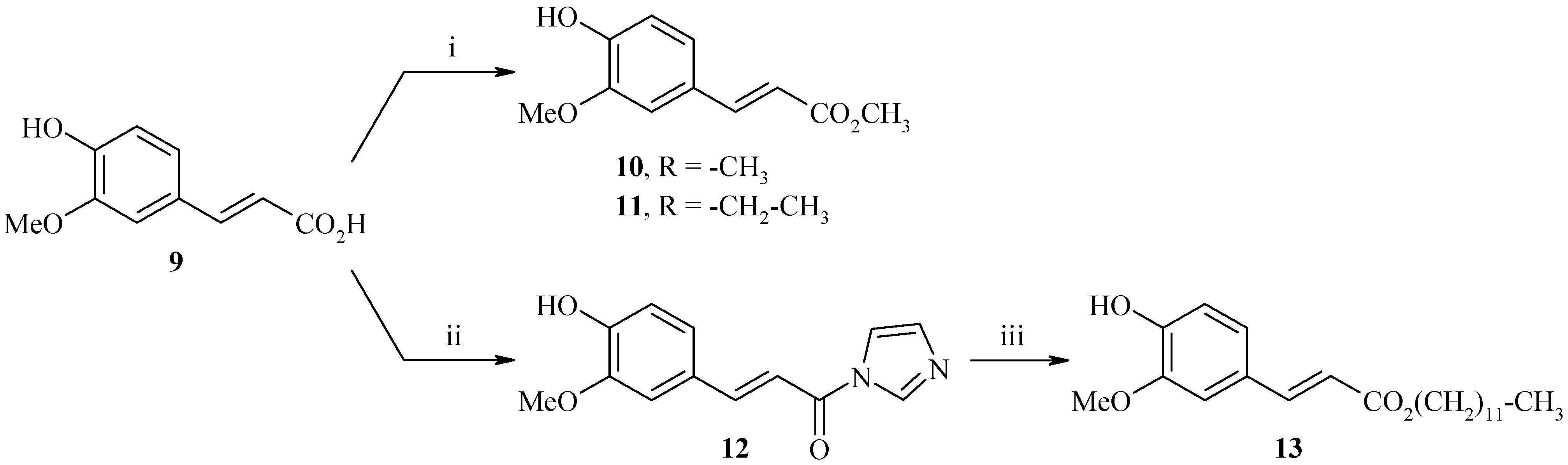
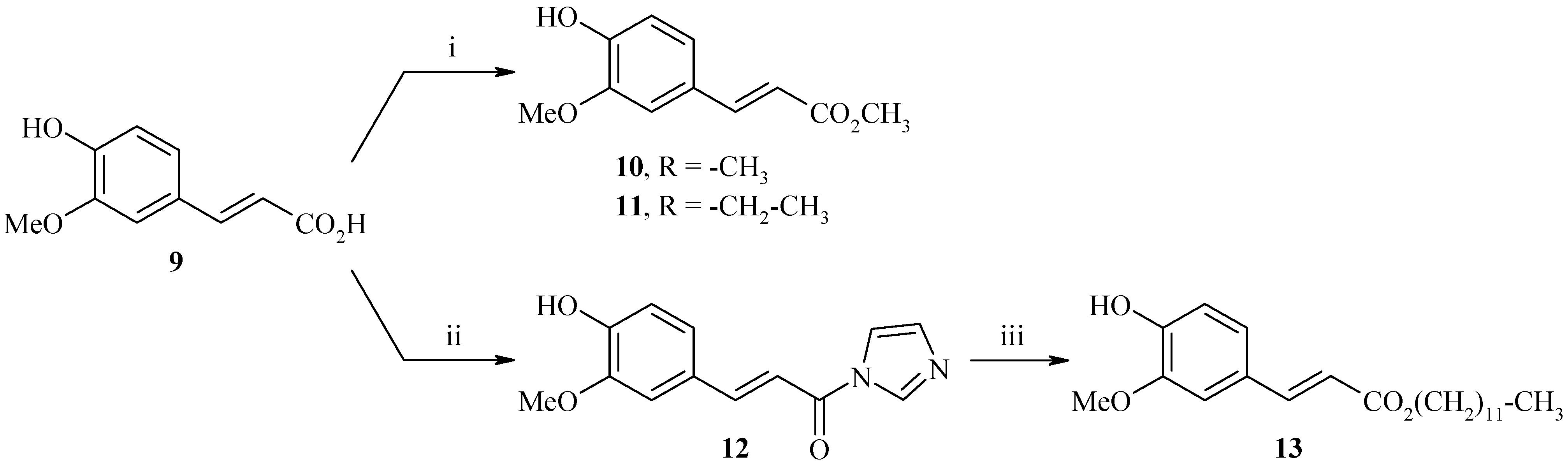
Synthesis of methyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (10)
Ferulic acid 9 (30.0 g, 154.0 mmol) and 5 drops of H2SO4 (95%) were refluxed in methanol for 24 h. The crude product was then evaporated to dryness and re-dissolved in dichloromethane. The organic layer was washed with saturated NaHCO3 solution (2 x 50 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane-ethylacetate: 8/2) to afford 31.2 g of methyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (10) as a white solid (yield: 97%); m.p.: 68°C; IR (cm-1): 3399 (OH), 2947 (CH3), 1718 (C=O), 1264-1160 (C-O); 1H-NMR: 9.62 (bs, 1H, OH), 7.55 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.31 (s, 1H, H2’), 7.11 (d, 3J=8.1 Hz, 1H, H6’), 6.78 (d, 3J=8.1 Hz, 1H, H5’), 6.47 (d, 3Jtrans=15.9 Hz, 1H, H3), 3.80 (s, 3H, CO2CH3), 3.68 (s, 3H, OCH3); 13C-NMR: 167.5, 149.8, 148.3, 145.5, 125.9, 123.5, 115.9, 114.6, 111.7, 53.1, 51.6; MS (ESI+) m/z 209 (M++1).
Synthesis of ethyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (11)
Ferulic acid 9 (50.0 g, 257.0 mmol) and 10 mL of H2SO4 (95%) were refluxed in ethanol for 24h. The crude product was then evaporated to dryness and re-dissolved in dichloromethane. The organic layer was washed with saturated NaHCO3 solution (2 x 50 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane-ethyl acetate: 8/2) to afford 45.8 g of ethyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (11) as a white solid (yield: 80%); m.p.: 67°C; IR (cm-1): 3180 (OH), 2952 (CH3), 1711 (C=O), 1256 (C-O); 1H-NMR: 9.58 (bs, 1H, OH), 7.53 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.31 (s, 1H, H2’), 7.10 (d, 3J=8.1 Hz, 1H, H6’), 6.77 (d, 3J=8.1 Hz, 1H, H5’), 6.45 (d, 3Jtrans=15.9 Hz, 1H, H3), 4.15 (q, J=7.0 Hz, 2H, CH2), 3.76 (s, 3H, OCH3), 1.23 (t, J=7.0 Hz, 3H, CH3); 13C-NMR: 166.9, 148.8, 148.3, 145.1, 126.9, 123.9, 116.5, 115.6, 110.7, 60.3, 56.4, 14.3; MS (ESI+) m/z 223 (M++1).
Synthesis of (E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1H-imidazol-1-yl)prop-2-en-1-one (12)
To a solution of ferulic acid (9, 5.0 g, 25.8 mmol) in freshly distilled THF (60 mL) was added 1,1'-carbonyldiimidazole (8.3 g, 51.5 mmol) and the mixture was refluxed for 3.5 h (CaCl2 guard tube). The resulting mixture was cooled down to room temperature and concentrated in vacuo. The residue was chromatographed on silica gel (ether) to afford 4.8 g of (E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1H-imidazol-1-yl)prop-2-en-1-one (12) as a white solid (yield: 76%); m.p.: 156°C; IR (cm-1): 3124 (OH), 2841 (CH3), 1772 (C=O), 1320-1243 (C-O); 1H-NMR: 8.78 (bs, 1H, Himidazole), 8.50 (bs, 1H, Himidazole), 8.03 (d, 3Jtrans=15.5 Hz, 1H, H2), 7.94 (s, 1H, H2’), 7.62 (d, 3J=8.0 Hz, 1H, H5’), 7.70 (d, 3Jtrans=15.5 Hz, 1H, H3), 7.55 (d, 3J=8.0 Hz, 1H, H6’), 7.17 (bs, 1H, Himidazole), 3.90 (s, 3H, OCH3); 13C-NMR: 163.3, 149.5, 147.8, 138.2, 129.9, 129.2, 128.2, 124.3, 115.5, 115.3, 111.8, 111.4, 57.0; MS (EI) m/z: 244 (M+ •, 100).
Synthesis of dodecyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (13)
To a solution of (E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1H-imidazol-1-yl)prop-2-en-1-one (12, 4.8 g, 19.7 mmol) in freshly distilled THF (60 mL) were added DBU (3.0 g, 19.7 mmol) and 1-dodecanol (3.7 g, 19.7 mmol) and the mixture was refluxed for 48 h. The resulting mixture was cooled down to room temperature and concentrated in vacuo. The crude product was diluted with water (40 mL) and extracted with ethyl acetate (3 x 50 mL). The organic layer was dried (MgSO4), and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane-ethyl acetate: 7/3) to afford 4.7 g of dodecyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (13) as a white solid (yield: 66%); m.p.: 46°C; IR (cm-1): 3534 (OH), 3131, 2923 (CH3), 2849, 1746 (C=O), 1269-1190 (C-O); 1H-NMR: 9.58 (bs, 1H, OH), 7.52 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.29 (s, 1H, H2’), 7.08 (d, 3J=8.1 Hz, 1H, H6’), 6.77 (d, 3J=8.1 Hz, 1H, H5’), 6.44 (d, 3Jtrans=15.9 Hz, 1H, H3), 4.09 (t, 3J=6.5 Hz, 2H, OCH2), 3.80 (s, 3H, OCH3), 1.60-1.58 (m, 2H, CH2), 1.31-1.21 (m, 18H, (CH2)9), 0.82 (t, 3J=6.4 Hz, 3H, CH3); 13C-NMR: 167.4, 147.8, 146.8, 144.6, 127.2, 123.0, 115.6, 114.6, 110.1, 64.6, 56.1, 31.9, 30.4, 29.8, 29.6, 29.5, 29.4, 29.3, 28.7, 25.9, 22.7, 15.1; MS (EI) m/z: 362 (M+ •, 95).
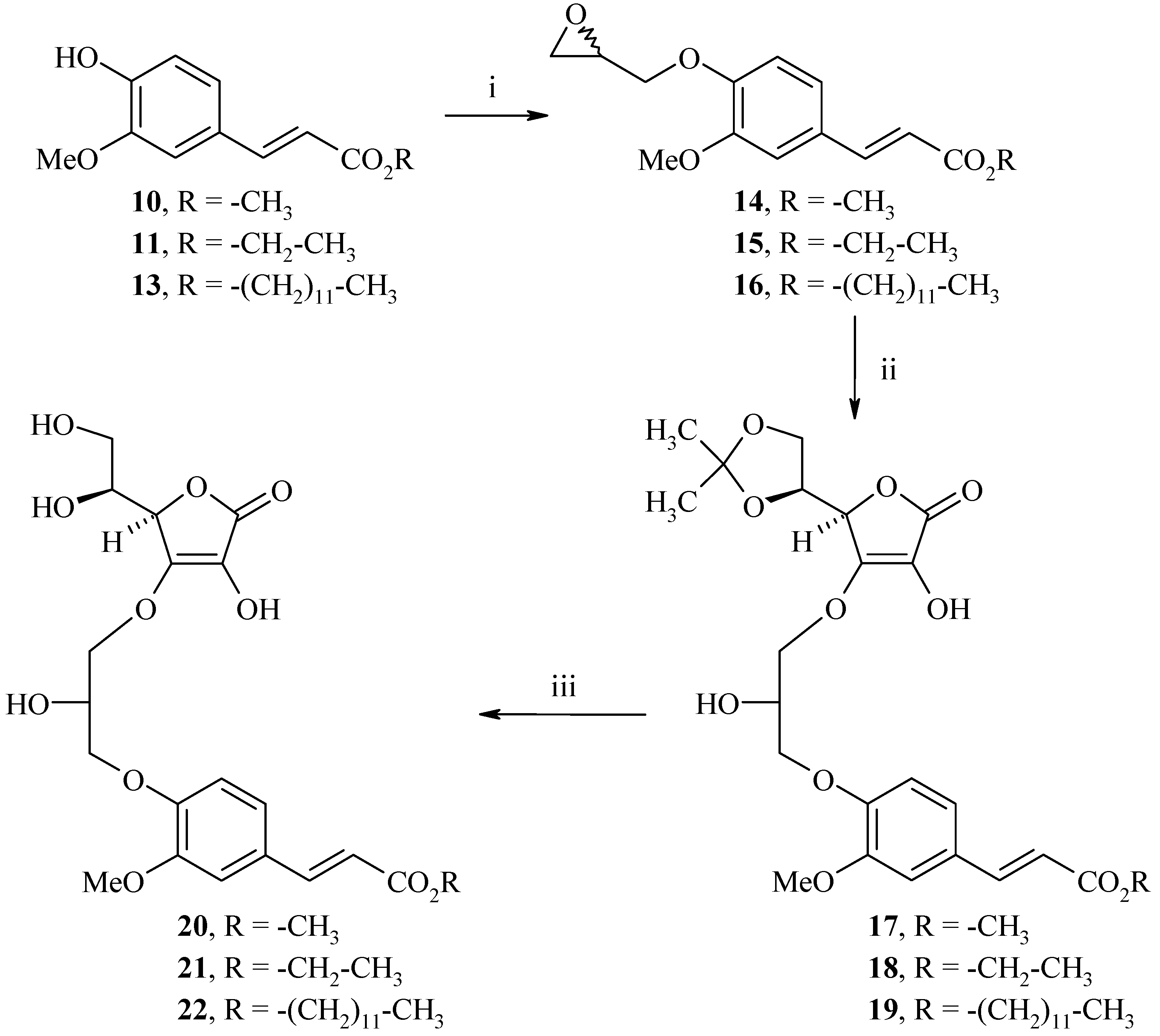
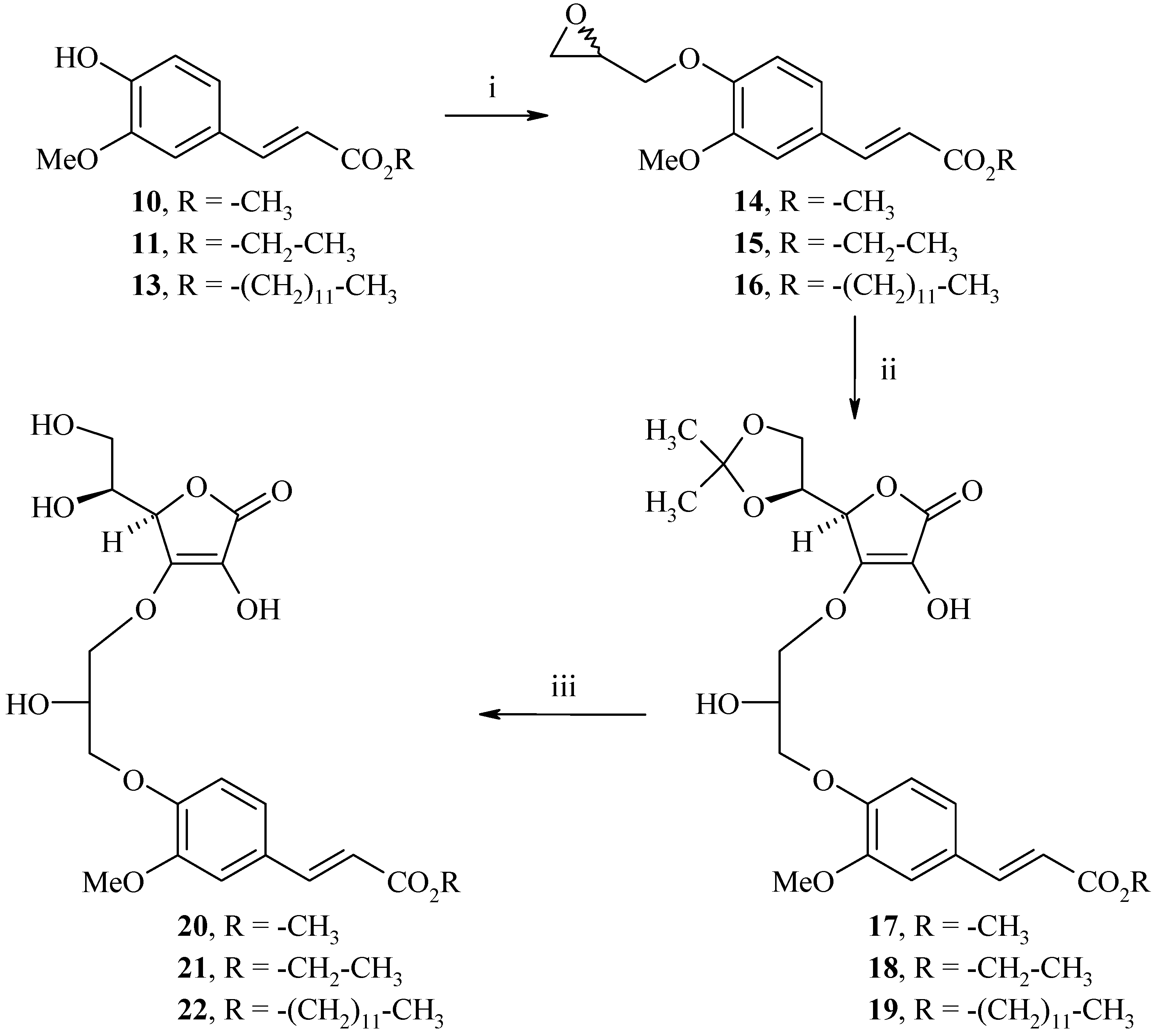
Synthesis of methyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (14)
To a water-acetone medium (1:1, 80 mL) were added methyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (10, 3.1 g, 15.0 mmol) and NaOH (0.6 g, 15.0 mmol). The mixture was stirred at room temperature for 10 min. (±)-Epichlorohydrin (12.8 g, 75.0 mmol) was added and the resulting mixture was refluxed for 8 h. Then, acetone was concentrated in vacuo and the aqueous layer was diluted with 70 mL of cold water and extracted with ethyl acetate (3 x 40 mL). The organic layer was dried (MgSO4), and concentrated in vacuo. Recrystallization from diethyl ether afford 2.8 g of methyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (14) as a white solid (yield: 70%); m.p.: 100°C; IR (cm-1): 2943-2925 (CH3), 2837 (CH2), 1717 (C=O), 1264-1161 (C-O); 1H-NMR: 7.58 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.37 (s, 1H, H2’), 7.22 (d, 3J=8.1 Hz, 1H, H6’), 6.98 (d, 3J=8.1 Hz, 1H, H5’), 6.57 (d, 3Jtrans=15.9 Hz, 1H, H3), 4.34-4.33 (m, 1H, Hoxirane), 3.81 (s, 3H, CO2CH3), 3.70 (s, 3H, OCH3), 3.33-3.32 (m, 2H, CH2), 2.84-2.83 (m, 1H, Hoxirane), 2.82-2.81 (m, 1H, Hoxirane); 13C-NMR: 167.9, 149.5, 147.8, 144.2, 128.8, 122.8, 117.5, 113.0, 111.3, 70.2, 55.7, 51.5, 49.8, 44.6; MS (EI) m/z: 264 (M+ •, 41), 179 (37), 15 (100).
Synthesis of ethyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (15)
The same procedure as compound 14 starting from ethyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (11, 20.0 g, 90.0 mmol) and (±)-epichlorohydrin (76.7 g, 450.0 mmol) was used to prepare 11.3 g of ethyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (15) as a white solid (yield: 45%); m.p.: 75°C; IR (cm-1): 2980 (C-H), 1703 (C=O), 1600 (C=C), 1309-1273 (C-O); 1H-NMR: 7.56 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.37 (s, 1H, H2’), 7.21 (d, 3J=8.2 Hz, 1H, H6’), 6.97 (d, 3J=8.2 Hz, 1H, H5’), 6.56 (d, 3Jtrans=15.9 Hz, 1H, H3), 4.34-4.33 (m, 1H, Hoxirane), 4,16 (q, J=7.0 Hz, 2H, CH2), 3.81 (s, 3H, OCH3), 3.33-3.32 (m, 2H, CH2), 2.84-2.83 (m, 1H, Hoxirane), 2.70-2.69 (m, 1H, Hoxirane), 1.24 (t, J=7.0 Hz, 3H, CH3); 13C-NMR: 166.9, 149.5, 147.8, 144.6, 128.3, 122.9, 116.6, 113.2, 111.3, 70.2, 60.3, 55.7, 51.5, 49.8, 44.6, 14.4; MS (ESI+) m/z 279 (M++1).
Synthesis of dodecyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (16)
The same procedure as compound 14 starting from dodecyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (13, 1.8 g, 5.0 mmol) and (±)-epichlorohydrin (2.3 g, 25.0 mmol) gave 1.7 g of dodecyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (16) as a white solid (yield: 83%); m.p.: 69°C; IR (cm-1): 2919-2849 (CH3, CH2), 1713 (C=O), 1265-1171 (C-O); 1H-NMR: 7.58 (d, 3Jtrans=16.0 Hz, 1H, H2), 7.38 (s, 1H, H2’), 7.22 (d, 3J=8.1 Hz, 1H, H6’), 6.98 (d, 3J=8.1 Hz, 1H, H5’), 6.57 (d, 3Jtrans=16.0 Hz, 1H, H3), 4.34-4.33 (m, 1H, Hoxirane), 4.12 (t, 3J=7.2 Hz, 2H, OCH2), 3.83 (s, 3H, OCH3),3.63-3.62 (m, 2H, CH2oxirane), 2.86-2.84 (m, 1H, Hoxirane), 2.71-2.69 (m, 1H, Hoxirane); 1.64-1.62 (m, 2H, CH2), 1.28-1.18 (m, 18H, (CH2)9), 0.86 (t, 3J=6.4 Hz, 3H, CH3); 13C-NMR: 167.4, 149.5, 147.8, 144.5, 128.2, 122.9, 115.6, 113.6, 111.3, 70.2, 64.6, 55.7, 49.8, 44.6, 31.9, 30.4, 29.8, 29.6, 29.5, 29.4, 29.3, 28.7, 25.9, 22.7, 14.1; MS (EI) m/z: 418 (M+ •, 37), 169 (100).
Synthesis of methyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (17)
To a solution of methyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (14, 2.2 g, 8.3 mmol) and (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8, 2.2 g, 10.0 mmol) in 1,4-dioxane (30 mL) were added NaHCO3 (0.8 g, 8.3 mmol) and 4-dimethylaminopyridine (0.1 g, 0.8 mmol). The resulting mixture was refluxed for 24 h. The organic layer was concentrated in vacuo and the crude product was diluted with water (100 mL) and extracted with ethyl acetate (3 x 40 mL). The organic layer was dried (MgSO4), and concentrated in vacuo. Recrystallization from diethyl ether afforded 2.0 g of methyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (17) as a white solid (yield: 50%); m.p.: 73°C; IR (cm-1): 3445 (OH), 2988-2950 (CH), 1760 (C=O), 1699 (C=C), 1260 (C(CH3)2); 1H-NMR: 9.03 (bs, 1H, OH), 7.58 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.35 (s, 1H, H2), 7.22 (d, 3J=8.1 Hz, 1H, H6), 6.99 (d, 3J=8.1 Hz, 1H, H5), 6.55 (d, 3Jtrans=15.9 Hz, 1H, H3), 5.43 (bs, 1H, OH), 4.78-4.77 (m, 1H, H4), 4.43-4.40 (m, 1H, H5), 4.21-4.20 (m, 1H, H6), 4.14-3.97 (m, 6H), 3.80 (s, 3H, CO2CH3), 3.70 (s, 3H, OCH3), 1.24-1.22 (m, 6H, CH3); 13C-NMR: 170.3, 167.5, 149.8, 149.2, 148.8, 148.6, 144.4, 127.9, 122.2, 119.6, 115.6, 113.1, 110.1, 74.9, 71.9, 69.9, 69.5, 68.4, 64.9, 55.6, 51.4, 25.5, 25.2; MS (EI) m/z: 480 (M+ •, 8), 280 (56), 206 (66), 194 (100).
Synthesis of ethyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (18)
The same procedure as compound 17 starting from ethyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (15, 2.2 g, 8.3 mmol) and (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8, 2.2 g, 10.0 mmol) was used to give 1.6 g of ethyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)- 3-methoxyphenyl)acrylate (18) as a white solid (yield: 49%); m.p.: 71°C; IR (cm-1): 3432 (OH), 2985 (CH), 1770 (C=O), 1698 (C=C), 1259 (C(CH3)2); 1H-NMR: 8.90 (bs, 1H, OH), 7.49 (d, 3Jtrans= 15.9 Hz, 1H, H2), 6.98 (s, 1H, H2), 6.96 (d, 3J=8.3 Hz, 1H, H6), 6.79 (d, 3J= 8.3 Hz, 1H, H5), 6.22 (d, 3Jtrans= 15.9 Hz, 1H, H3), 4.58 (bs, 1H, OH), 4.51 (m, 1H, H4), 4.30 (m, 1H, H5), 4.22 (m, 1H, H6), 4,16 (q, J=7.0 Hz, 2H, CH2), 4.10-3.86 (m, 6H), 3.78 (s, 3H, OCH3), 1.35-1.33 (m, 3H, CH3), 1.27-1.23 (m, 6H, CH3); 13C-NMR: 174.4, 172.2, 166.9, 149.3, 146.5, 144.5, 128.3, 126.7, 123.1, 116.6, 112.8, 111.4, 110.6, 77.7, 76.1, 72.0, 70.4, 70.3, 65.1, 60.3, 55.7, 26.4, 26.0, 14.4; MS (ESI+) m/z 495 (M++1).
Synthesis of dodecyl (2E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (19)
The same procedure as compound 17 starting from dodecyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (16, 1.7 g, 4.1 mmol) and (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8, 1.1 g, 5.0 mmol) gave 1.7 g of dodecyl (2E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxy-propoxy)-3-methoxyphenyl)acrylate (19) as a colourless oil (yield: 66%); IR (cm-1): 3440 (OH), 2924-2854 (CH), 1769 (C=O), 1700 (C=C), 1260 (C(CH3)2); 1H-NMR: 9.03 (bs, 1H, OH), 7.56 (d, 3Jtrans=16.0 Hz, 1H, H2), 7.35 (s, 1H, H2), 7.20 (d, 3J=7.8 Hz, 1H, H6), 6.98 (d, 3J=7.8 Hz, 1H, H5), 6.55 (d, 3Jtrans=16.0 Hz, 1H, H3), 5.43 (bs, 1H, OH), 4.77-4.76 (m, 1H, H4), 4.43-4.39 (m, 2H), 4.21-4.20 (m, 1H, H6), 4.14-3.97 (m, 5H), 4.10-4.09 (m, 2H, OCH2), 3.80 (s, 3H, OCH3), 1.64-1.62 (m, 2H, CH2), 1.24 (s, 6H, CH3), 1.23-1.21 (m, 18H, (CH2)9), 0.84-0.82 (m, 3H, CH3); 13C-NMR: 174.4, 172.3, 167.4, 149.3, 146.5, 144.5, 128.2, 126.6, 123.1, 115.7, 112.8, 111.5, 110.7, 77.7, 76.1, 70.5, 72.0, 70.1, 65.1, 64.6, 53.7, 31.9, 30.4, 29.8, 29.6, 29.5, 29.4, 29.3, 28.7, 26.3, 26.1, 25.9, 22.7, 14.1; MS (EI) m/z: 635 (M+ •, 5), 436 (69), 362 (65), 194 (100).
Synthesis of methyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (20)
To a solution of methyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (17, 2.0 g, 4.2 mmol) in 1,4-dioxane (30 mL) was added HCl (12 N, 0.6 mL) and the resulting mixture was stirred at room temperature for 4 h. The organic layer was neutralized with saturated NaHCO3 solution (100 mL), and extracted with ethyl acetate (3 x 40 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed on silica gel (ethyl acetate) to afford 1.3 g of methyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)-acrylate (20) as a white solid (yield: 71%); m.p.: 69°C; [α = +21.0 (c 0.5, C2H5OH); IR (cm-1): 3452 (OH), 2953 (CH), 1765 (C=O), 1692 (C=C), 1259 (C-O); 1H-NMR: 8.82 (bs, 1H, OH), 7.58 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.35 (s, 1H, H2), 7.22 (d, 3J=8.1 Hz, 1H, H6), 7.00 (d, 3J=8.1 Hz, 1H, H5), 6.56 (d, 3Jtrans=15.9 Hz, 1H, H3), 5.43 (bs, 1H, OH), 4.96-4.78 (m, 2H, H4, H5), 4.43-4.41 (m, 2H, H6), 4.00-3.99 (m, 5H), 3.80 (s, 3H, CO2CH3), 3.70 (s, 3H, OCH3); 13C-NMR: 170.5, 167.0, 150.41 149.2, 144.7, 127.2, 122.9, 119.3, 118.1, 115.4, 112.9, 110.8, 74.6, 71.9, 69.7, 68.6, 67.6, 61.7, 55.7, 51.3. MS (ESI+) m/z 441 (M++1).
\Synthesis of ethyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (21)
The same procedure as compound 20 starting from ethyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)-acrylate (18, 0.3 g, 0.5 mmol) was used to prepare 8.6 mg of ethyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)-acrylate (21) as a white solid (yield: 61%); m.p.: 68°C; [α = +21.0 (c 0.5, C2H5OH); IR (cm-1): 3419 (OH), 2936 (CH), 1760 (C=O), 1694 (C=C), 1259 (C-O); 1H-NMR: 8.83 (bs, 1H, OH), 7.56 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.34 (s, 1H, H2), 7.20 (d, 3J=8.0 Hz, 1H, H6), 6.99 (d, 3J=8.0 Hz, 1H, H5), 6.53 (d, 3Jtrans=15.9 Hz, 1H, H3), 5.44 (bs, 1H, OH), 4.97-4.38 (m, 2H, H4, H5), 4.18-4.00 (m, 5H), 3.80 (s, 3H, OCH3), 1.23 (t, J=7.0 Hz, 3H, CH3); 13C-NMR: 170.9, 166.0, 150.7, 149.6, 144.9, 127.7, 123.2, 119.8, 118.7, 116.2, 113.4, 111.2, 75.0, 72.3, 70.3, 69.0, 67.9, 62.2, 60.2, 56.1, 14.5; MS (ESI+) m/z 455 (M++1).
Synthesis of dodecyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (22)
The same procedure as compound 20 starting from dodecyl (2E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)-acrylate (19, 0.4 g, 0.6 mmol) was used to prepare 0.3 g of dodecyl (2E)-3-[4-(3-[2-(1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydro-furan-3-yloxy]-2-hydroxy-propoxy)3-methoxyphenyl]-acrylate (22) as a white solid (yield: 75%); m.p.: 67°C; [α = +21.6 (c 0.5, C2H5OH); IR (cm-1): 3387 (OH), 2953-2853 (CH), 1759 (C=O), 1695 (C=C), 1260 (C-O); 1H-NMR: 8.83 (bs, 1H, OH), 7.56 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.35 (s, 1H, H2), 7.20 (d, 3J=8;1 Hz, 1H, H6), 6.99 (d, 3J=8.1 Hz, 1H, H5), 6.54 (d, 3Jtrans=15.9 Hz, 1H, H3), 5.42 (bs, 1H, OH), 4.95-4.78 (m, 2H, H4, H5), 4.46-4.45 (m, 2H, H6), 4.12-4.01 (m, 5H), 3.81 (s, 3H, OCH3), 1.61-1.60 (m, 2H, CH2), 1.22-1.19 (m, 20H, (CH2)10), 0.83 (t, 2J=5.8 Hz, 3H, CH3); 13C-NMR: 170.5, 166.6, 150.5, 150.2, 149.2, 134.9, 127.2, 122.8, 119.4, 115.6, 112.7, 110.6, 74.6, 69.7, 68.6, 67.5, 66.4, 63.8, 61.7, 55.7, 31.3, 30.4, 29.0, 28.9, 28.8, 28.7, 28.6, 28.3, 25.4, 22.1, 13.9; MS (ESI+) m/z 596 (M++1).

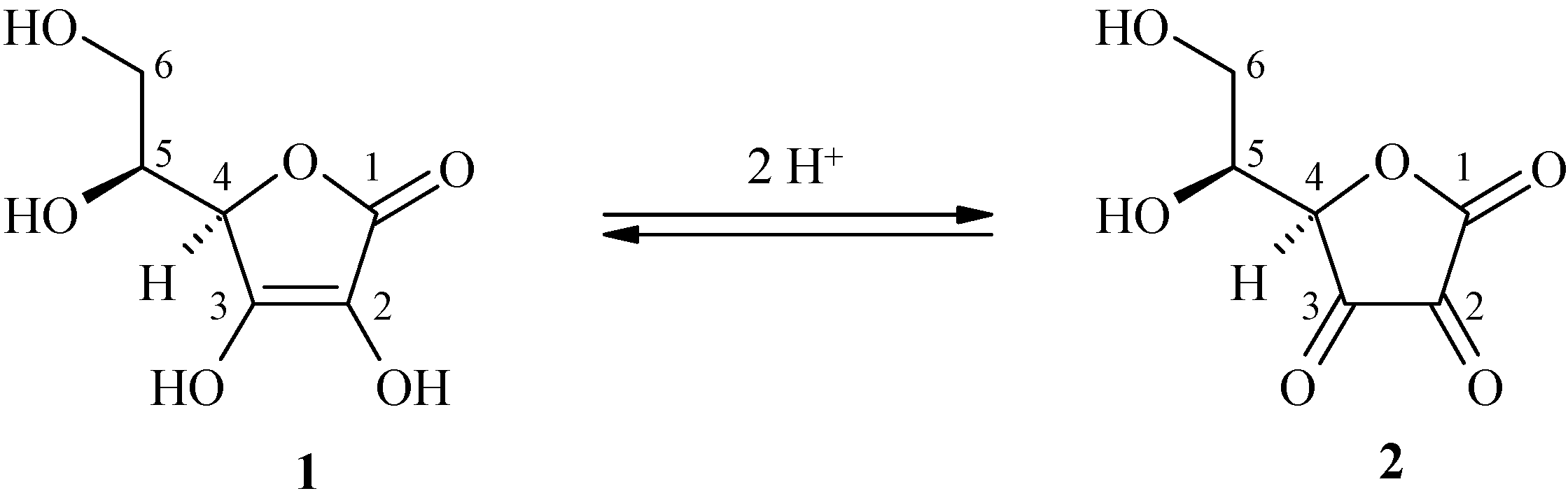


{kind=link}
{kind=link}
{kind=link}
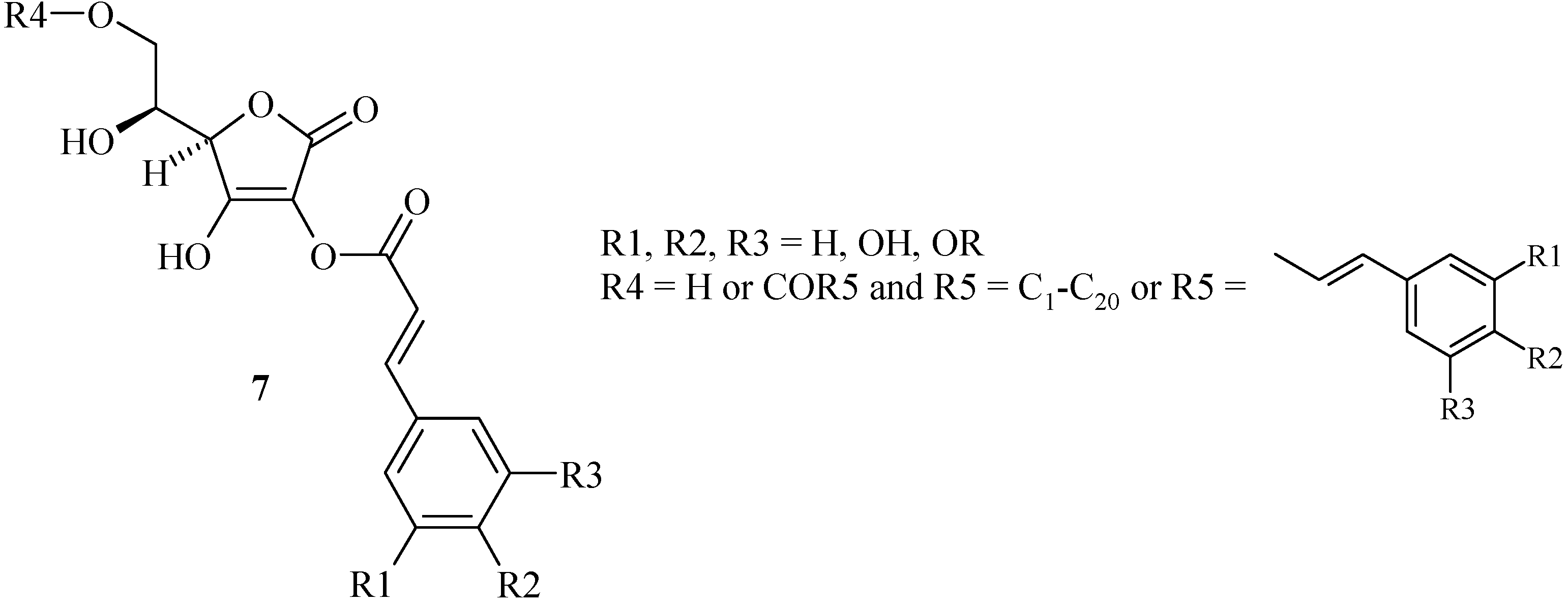{kind=link}
{kind=link}
{kind=link}
{kind=link}