Results and Discussion
Our main goal in the oxidative degradation of the
ent-labdane side chains was to obtain compounds
5 and
7 (
Scheme 1), and use these to generate a primary alcohol at the C-12 position. This primary alcohol would then be transformed into a good leaving group, which in turn would lead to suitable terpenic fragments that could be reacted with quinone/hydroquinone moieties in an attempt to synthesize terpenylquinones/hydroquinones with potential anticancerogenic activities. The preparation of the primary alcohols and the synthesis of the terpenylquinones/hydroquinones will be reported elsewhere.
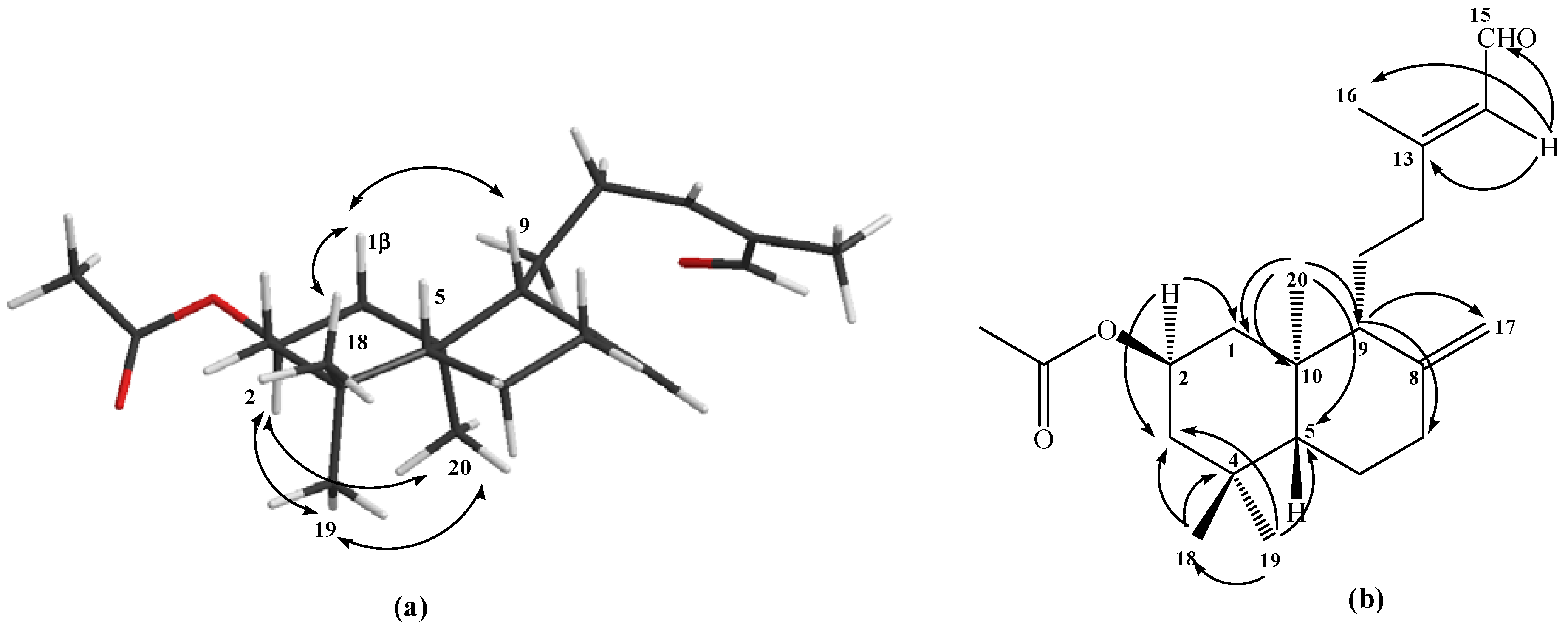
The structural determination of the mixture of compounds
1-2 was accomplished by
1H- and
13C-NMR techniques. The spectral data, when compared with those previously reported for compounds with similar structures containing
ent-labdane skeletons [
21,
22], were consistent with a diterpenic bicyclic labdane structure having a Δ
8(17) double bond and a 2β-hydroxyl group. Examination of the
13C-NMR spectra of acetylated derivates
1a-2a, showed that they were a 60/40 mixture of the C-13
E and
Z isomers, respectively. After successive purifications of
1a-2a by column chromatography (C.C.) a 10:1
E:Z mixture was isolated (ratio calculated based on the integrals of the Me-16 signals in the
1H-NMR spectrum). This mixture was used for the structural determination of compound
1a, whose
1H-NMR spectrum indicated the existence of a hydrogen at
δ 9.96 (1H, d,
J=8.1Hz, H-15) corresponding to an aldehyde function, three singlet methyl groups at
δ 0.75, 0.86 and 0.91 ppm corresponding to Me-18, Me-19 and Me-20, respectively, as well as two broad singlets at
δ 4.49 and 4.87 ppm for the geminal H-17 vinylic protons. A double double triplet at
δ 4.99 ppm was also assignable to the hydrogen geminal to an acetyl group (C
H3CO
δ 2.00 ppm), and with the aid of a gs-sel-
1H 1D TOCSY experiment, it was unequivocally determined to be located between C-1 and C-3. No NOE cross peak was observed between H-5 and Me-20, which suggested that the latter is oriented
trans to H-5. The relative stereochemistry of
1a was assigned on the basis of both a NOESY correlation and coupling constant data. Main NOESY correlations observed for
1a are shown in
Figure 1, which indicated that ring
A and
B were trans-fused and H-2 was axially oriented. On the other hand, an axial-axial (
Jaa=11.8 Hz) and axial-equatorial (
Jae=4.2 Hz) coupling constant for H-1, H-2α and H‑3 indicated that the OAc group in C-2 was
β-equatorial (see
Figure 1a). Furthermore, an allylic methyl group, as indicated by a signal in the
1H-NMR spectrum at
δ 2.16 ppm (C-16,
δ 17.6 ppm), and long range coupled to a broad singlet corresponding to a vinylic proton at
δ 5.86 ppm (see
Figure 1b) were characteristic of an unsaturated side chain of a bicyclic diterpenoid having an
E configuration at the olefinic bond [
23]. Nevertheless, since our plans called for the loss of isomeric relationships at C-13, the
1-2 mixture was used directly as our synthesis.
Figure 1.
Structure of compound 1a. (a) NOE correlations. (b) HMBC correlations.
Figure 1.
Structure of compound 1a. (a) NOE correlations. (b) HMBC correlations.
Acetylation of the 1-2 mixture under standard conditions (Ac2O/pyridine/DMAP/CH2Cl2) produced the acetate mixture 1a-2a in 98% yield. Reduction of this acetate mixture with NaBH4/CH3OH after C.C. purification led to the formation of the geometrical isomers 3 (28%) and 4 (43%). The confirmation of the E and Z structures of these geometrically isomeric alcohols, was made by 1H- and 13C-NMR data; specifically the E isomer 4 showed a carbinol methylene group at δ = 4.13 (2H, d, J=6.9Hz, H-15) and a signal at δ = 59.3 (C-15) in its 13C spectrum, whereas the Z isomer 3 showed a carbinol methylene group at δ = 4.02 (2H, dd, J=6.9 and 4.2Hz, H-15) and a 13C signal at δ = 58.9 (C-15). A singlet peak at δ = 1.65 (3H, s, H-16) and δ = 16.3 (C-16) indicated that the stereochemistry of the double bond in the side chain corresponded to E. In the case of the Z isomer, the Me-16 appears at δH = 1.70 and δC = 23.3 ppm, respectively.
Degradation of the side chain of the mixture of compounds
1a-2a using either the OsO
4/NaIO
4 or RuCl
3/NaIO
4 systems, a degradation used previously for compounds with similar structures [
1,
2,
3,
14], led to the formation of compounds
5 and
6 (
Scheme 1). Nevertheless, when the mixture
1a-2a was treated with the RuCl
3/NaIO
4 system, a greater yield of compound
5 was obtained (76% v/s 68% with OsO
4/NaIO
4 system). The main spectroscopic data for the confirmation of the structure of
5 was the appearance of a singlet signal at
δH = 2.05 (3H,
s, H-16) and
δC = 29.8 (C-16); in addition, in its
13C spectrum, the presence of two ketone carbonyl signals was observed at
δC = 211.3 (C-8) and 208.9 (C-13). The molecular formula for compound
6 (C
20H
32O
4) was deduced from the combined spectra of
1H,
13C,
13C-DEPT-135 and confirmed by MS ([M
+ 336] at m/z).
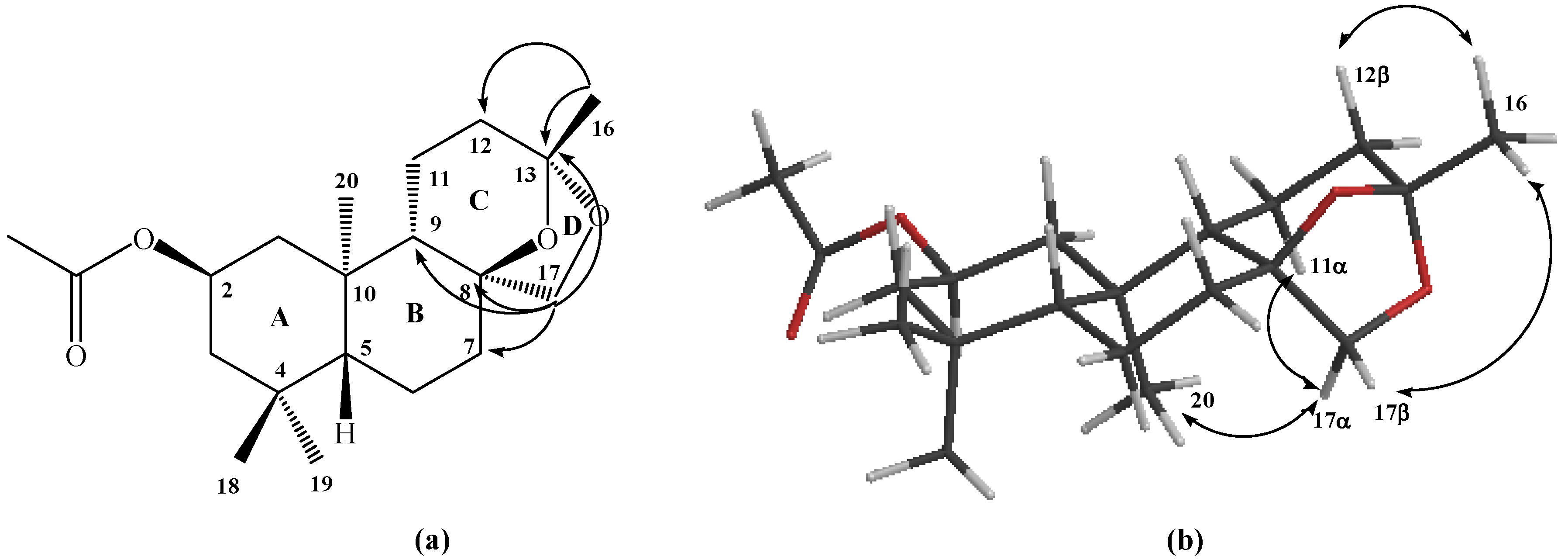
Our calculation of the degree of unsaturation gave a value of five, of which one corresponds to the acetate group, and two are attributed to the A and B rings. In the 13C spectrum, no signals corresponding to C=C double bonds were observed, therefore the formation of a third ring C fused with B, formed by the union between the carbons C-8, C-9, C-11, C-12, C-13-O-C-8, and a fourth ring D fused with C, formed by an ether bridge between C-13 (ketal) and C-17 is suggested.
The tetracyclic structure of compound
6 was confirmed mainly by the data obtained from heteronuclear 2D HSQC and HMBC correlations; in the latter, correlations were observed between H‑17β with C-7, C-8 and C-9, whereas H-17α shows correlations with C-9 and C-13, while the Me-16 group also showed correlations with C-13 and C-12 (see
Figure 2a). The stereochemistry assigned to the carbons C-8 and C-13 were deduced from
1H sel. 1D NOESY correlations; long range interactions between the Me-16 group with H-12β and H-17β were observed, whereas H-17α showed correlations with Me-20 and H-11α (see
Figure 2b). Finally, a tetracyclic structure similar to
6 was reported when labdadienes were treated with O
3 or NaIO
4/OsO
4 system [
14].
It is reported that allylic alcohols like (-)-sclareol or lab-13-en-8β-ol-15-oic acid (α,β-unsaturated methyl esters) were selectively oxidized to the corresponding methyl ketones with the KMnO
4/MgSO
4 in acetone system [
24,
25]. Under these conditions, when compounds
3,
4 or a mixture of both were treated with KMnO
4/MgSO
4 in acetone at room temperature, compound
7 was obtained selectively in 63% yield. Specifically, in the
1H-NMR spectrum of
7, the Me-16 appears at
δH = 2.03 (s, 3H), whereas in the
13C spectrum, the CH
3-CO group showed signals at
δC = 29.9 (C-16) and 208.8 ppm (C-13). On the other hand, the exocyclic double bond between C-8 and C-17 showed a signal at
δH = 4.79 (1H,
bs, H-17a) and
δH = 4.40 (1H,
bs, H-17b),
δC = 147.0 (C-8) and 107.2 (C-17). Combined, all these signals confirm the structure of compound
7 and with it, the selectivity of the side chain oxidation reaction using KMnO
4.
Figure 2.
Structure of compound 6. (a) HMBC correlations. (b) NOE correlations.
Figure 2.
Structure of compound 6. (a) HMBC correlations. (b) NOE correlations.
Experimental
General
Unless otherwise stated, all chemical reagents purchased (Merck or Aldrich) were the highest purity commercially available and were used without previous purification. IR spectra were recorded as thin films in a Nicolet Impact 420 spectrometer and frequencies are reported in cm-1. Optical rotations were measured with a sodium lamp (λ=589 nm, D line) on a Perkin Elmer 241 digital polarimeter equipped with 1 dm cells at the temperature indicated in each case. Low resolution mass spectra were recorded on a Shimadzu QP-2000 spectrometer at 70eV ionising voltage and are given as m/z (% rel. int.) 1H-, 13C- (DEPT 135 and DEPT 90), sel. 1D 1H NOESY, sel. 1D 1H TOCSY, 2D HSQC and 2D HMBC spectra were recorded in CDCl3 solutions and are referenced to the residual peaks of CHCl3 at δ 7.26 ppm and δ 77.0 ppm for 1H- and 13C-, respectively, on a Bruker Avance 400 Digital NMR spectrometer, operating at 400.1MHz for 1H and 100.6MHz for 13C. Chemical shifts are reported in δ ppm and coupling constants (J) are given in Hz. Silica gel (Merck 200-300 mesh) was used for C.C. and silica gel plates HF-254 for TLC. TLC spots were detected by heating after spraying with 25% H2SO4 in H2O.
Plant material
Aerial parts of Calceolaria inamoena Phil. were collected in Arica, Region I, Chile, in January 2006, and authenticated by Professor Eliana Belmonte, Universidad de Tarapacá. A voucher specimen (#98007) was deposited in the Herbarium of the Natural Product Laboratory of the Universidad Técnica Federico Santa María.
Extraction and isolation
Aerial parts of C. inamoena, (2,448 g of dry plant material) were successively extracted at room temperature with three 6.5 L portions of petroleum ether (b.p. 60-80°C) for 72 h each. The solvent was removed in vacuo to yield 110.28 g of a syrupy residue. The crude material was divided into two portions (approx. 55 g each), dissolved in CH2Cl2 and absorbed on silica. Both portions were then each subjected separately to silica gel column chromatography (450 g) eluting with mixtures of petroleum ether and EtOAc of increasing polarity (195:5→50:150). Fractions (47 frs., 10 mL, from a total of 248) were combined based on TLC and 1H-NMR monitoring. After purification by C.C. this produced 53.7 g of the geometric isomer mixture 1-2 (4.5% yield based on dry plant material).
Acetylation of a mixture of 1-2: synthesis of 2β-acetoxy-ent-labda-8(17),13(E)-dien-15-al (1a) and 2β-acetoxy-ent-labda-8(17),13(Z)-dien-15-al (2a).
To a solution of 1-2 mixture (30 g, 98.5 mmol) in dry CH2Cl2 (150 mL), dry pyridine (10 mL), DMAP (70 mg) and Ac2O (9.5 mL) were added and the mixture was stirred at room temperature for 2 h. To this mixture a cooled solution of 10% KHSO4 (approx. 25mL) was then added. The watery layer was discarded and the combined organic layers were washed to neutrality with a saturated solution of NaHCO3 and water, dried over Na2SO4, filtered, evaporated and chromatographed on silica-gel with petroleum ether/EtOAc mixtures of increasing polarity (19.8:0.2→5:15) to give a mixture of 1a-2a (33.4 g, 98%). Compound 1a: viscous oil, 1H-NMR: 9.96 (1H, d, J=8.1Hz, H-15), 5.86 (1H, d, J=8.1Hz, H-14), 4.99 (1H, ddt, J=12.0, 11.8 and 4.2Hz, H-2), 4.87 (1H, br, H-17b), 4.49 (1H, bs, H-17a), 2.16 (3H, s, Me-16), 2.00 (3H, s, OAc), 0.91 (3H, s, Me-18), 0.86 (3H, s, Me-19), 0.75 (3H, s, Me-20); 13C-NMR: 44.0 (C-1), 69.1 (C-2), 46.7 (C-3), 34.9 (C-4), 54.8 (C-5), 23.8 (C-6), 37.8 (C-7), 146.9 (C-8), 56.0 (C-9), 40.9 (C-10), 21.5 (C-11), 39.3 (C-12), 164.5 (C-13), 127.2 (C-14), 191.3 (C-15), 17.6 (C-16), 107.4 (C-17), 33.4 (C-18), 22.3 (C-19), 15.2 (C-20), 170.6 (CH3CO), 21.4 (CH3CO); IR: 2900-2980, 2850, 1720, 1640, 1460, 1395, 1360, 1250, 1140, 1020, 960, 895; MS: 346 ([M+] <1%), 289 (2.0%), 286 (5.3%), 203 (15.1%), 201 (10.5%), 189 (16.9%), 187 (15.7%), 175 (10.2%), 159 (20.3%), 157 (11.1%), 149 (11.7%), 147 (30.3%), 145 (23.8%), 136 (14.2%), 135 (100%), 133 (34.0%) 121 (30.7%). Compound 1b: 13C-NMR: 43.9 (C-1), 69.3 (C-2), 46.7 (C-3), 34.9 (C-4), 54.9 (C-5), 23.8 (C-6), 37.9 (C-7), 146.9 (C-8), 55.6 (C-9), 40.8 (C-10), 21.4 (C-11), 30.9 (C-12), 164.5 (C-13), 129.0 (C-14), 190.9 (C-15), 24.8 (C-16), 107.6 (C-17), 33.5 (C-18), 22.3 (C-19), 15.1 (C-20), 170.6 (CH3CO), 21.4 (CH3CO).
Reduction of the mixture of 1a-2a: synthesis of 2β-acetoxy-15-hydroxy-ent-labda-8(17),13(Z)-diene (3) and 2β-acetoxy-15-hydroxy-ent-labda-8(17),13(E)-diene (4)
NaBH4 (10.1g, 0.266 mol) was added in portions to a solution of 1a-2a mixture (10 g, 28.9 mmol) in MeOH (100 mL), and the mixture stirred at 10°C. After 2 h, and in order to destroy the excess of NaBH4, acetone (20 mL) was added and the mixture stirred at room temperature for 15 min. The solvent was removed until a volume of approximately 10 mL was reached and HCl (5%, 30 mL) was added and the mixture extracted with EtOAc (3 x 30 mL) and the combined organic layers washed successively with 10% NaHCO3 and water, dried over Na2SO4, filtered, evaporated and chromatographed on silica-gel with mixtures of petroleum ether/EtOAc of increasing polarity (19:1→6:14) to give 2.20 g (28%) of 3, 3.37 g (43%) of 4 and 2.28 g (29%) of 3+4 mixture, for a total yield of 7.85 g (78%). Compound 3: viscous oil; 1H-NMR: 5.39 (1H, brt, J=6.9Hz, H-14), 4.98 (1H, ddt, J=11.8, 11.8, 4.1 Hz, H-2), 4.88 (1H, s, H-17b), 4.56 (1H, s, H-17a), 4.02 (2H, dd, J=6.9, 4.2Hz, H2-15), 2.38 (1H, ddd, J=13.0, 3.9 and 2.3Hz, H-7α), 1.99 (1H, m, H-12b), 2.02 (1H, m, H-1α), 2.01 (3H, s, OAc), 1.99 (1H, m, H-12a), 1.93 (1H, ddd, J=13.0, 13.0 and 4.9Hz, H-7β), 1.72 (1H, m, H-3α), 1.70 (3H, s, Me-16), 1.69 (1H, m, H-6β), 1.59 (1H, brd, J=11.3Hz, H-9), 0.91 (3H, s, Me-18), 0.86 (3H, s, Me-19), 0.73 (3H, s, Me-20); 13C-NMR: 43.9 (C-1), 69.3 (C-2), 46.7 (C-3), 34.9 (C-4), 54.9 (C-5), 23.8 (C-6), 37.9 (C-7), 147.3 (C-8), 55.6 (C-9), 40.8 (C-10), 21.7 (C-11), 30.3 (C-12), 139.9 (C-13), 124.7 (C-14), 58.9 (C-15), 23.3 (C-16), 107.4 (C-17), 33.5 (C-18), 22.4 (C-19), 15.2 (C-20), 170.6 (CH3CO), 21.4 (CH3CO); IR: 3451, 3077, 2930, 1730, 1642, 1441, 1369, 1248, 1021; MS: 348 ([M]+ <1%), 288 (16.1%), 273 (72.1%), 225 (55.2%), 202 (33.3%), 175 (53.9%), 135 (86.7%), 121 (67.9%), 55 (66.1%), 41 (100%). Compound 4, viscous oil; 1H-NMR: 5.37 (1H, brt, J=6.8Hz, H-14), 5.00 (1H, ddt, J=11.8, 11.8 and 4.1Hz, H-2), 4.86 (1H, s, H-17b), 4.53 (1H, s, H-17a), 4.13 (2H, d, J=6.9Hz, H2-15), 2.38 (1H, ddd, J=12.9, 3.6 and 2.2Hz, H-7α), 2.14 (1H, ddd, J=14.0, 10.4 and 3.9Hz, H-12b), 2.04 (1H, m, H-1α), 2.02 (3H, s, OAc), 1.95 (1H, ddd, J=13.3, 12.9 and 4.8Hz, H-7β), 1.97 (1H, m, H-12a), 1.72 (1H, m, H-3α), 1.65 (3H, s, Me-16), 1.69 (1H, m, H-6β), 1.62 (1H, brd, J=11.1Hz, H-9), 0.92 (3H, s, Me-18), 0.87 (3H, s, Me-19), 0.75 (3H, s, Me-20); 13C-NMR: 44.1 (C-1), 69.3 (C-2), 46.8 (C-3), 34.9 (C-4), 54.9 (C-5), 23.8 (C-6), 37.9 (C-7), 147.3 (C-8), 56.2 (C-9), 40.9 (C-10), 22.0 (C-11), 38.3 (C-12), 140.1 (C-13), 123.2 (C-14), 59.3 (C-15), 16.3 (C-16), 107.3 (C-17), 33.5 (C-18), 22.4 (C-19), 15.2 (C-20), 170.4 (CH3CO), 21.3 (CH3CO); IR: 3380, 3068, 2945, 1725, 1640, 1445, 1373, 1251, 1028; MS: 348 ([M]+ <1%), 288 (16.1%), 273 (72.1%), 255 (55.2%), 187 (59.4%), 175 (53.9%), 135 (86.7%), 121 (67.9%), 41 (100%).
Oxidative degradation of a 1a-2a mixture with the OsO4-NaIO4 system: synthesis of 2β-acetoxy-14,15, 17-trinor-ent-labdane-8,13-dione (5) and 2β-acetoxy-(13R)-8β,13:13,17-diepoxy-14,15-dinor-ent-labdane (6)
Finely divided NaIO4 (3.1g, 14.5 mmol), was added to a solution of 1a-2a mixture (5g, 14.45 mmol) in 1:1 THF/H2O (80 mL) and the mixture stirred at room temperature under a N2 atmosphere. After 1 h, 4% aq. OsO4 solution (5 mL, 0.787 mmol) was added. The reaction mixture was then stirred at room temperature for 72 h, filtered and the solvent was removed until the volume was reduced to approximately 40 mL; then 5% Na2S2O5 (15 mL) was added, the mixture was extracted with EtOAc (3 x 30 mL) and the combined organic layers were washed successively with 10% NaHCO3 and water, dried over Na2SO4, filtered, evaporated and chromatographed eluting with mixtures of petroleum ether/EtOAc of increasing polarity (19.8:0.2→5:15) to give 3.16 g of compound 5 (68%) and 0.335 g of 6 (7.2%). Compound 5: viscous oil; [α]D23 = +21.1° (c 1.2, CHCl3); 1H-NMR: 4.93 (1H, ddt, J=12.1, 12.1 and 4.0 Hz, H-2), 2.56 (1H, ddd, J=17.7, 7.7 and 5.4Hz, H-12b), 2.39 (1H, ddd, J=13.6, 4.6 and 1.7Hz, H-7α), 2.26 (1H, dd, J=12.8, 7.0Hz, H-7β), 2.11 (1H, brd, J=10.7Hz, H-9), 2.05 (3H, s, Me-16), 1.99 (3H, s, OAc), 1.99 (1H, m, H-12a), 1.72 (1H, m, H-3α), 1.69 (1H, m, H-6β), 0.98 (3H, s, Me-18), 0.90 (3H, s, Me-19), 0.77 (3H, s, Me-20); 13C NMR: 43.8 (C-1), 68.3 (C-2), 46.4 (C-3), 34.8 (C-4), 53.4 (C-5), 23.0 (C-6), 42.1 (C-7), 211.3 (C-8), 62.7 (C-9), 43.6 (C-10), 16.2 (C-11), 42.3 (C-12), 208.9 (C-13), 29.8 (C-16), 33.5 (C-18), 22.3 (C-19), 15.3 (C-20), 170.4 (CH3CO), 21.3 (CH3CO). IR: 2950, 1738, 1723, 1721, 1429, 1368, 1245, 1189, 1025. MS: 322 ([M]+ 6.1%), 249 (12.1%), 247 (100%), 189 (34.5%), 180 (34.2%), 177 (29.7%), 135 (93.9%), 119 (38.2%), 43 (77.6%). Compound 6: viscous oil; [α]D23 = +2.9°(c 2.6, CHCl3); 1H-NMR: 5.02 (1H, ddt, J=12.2, 12.0 and 4.4 Hz, H-2), 4.25 (1H, d, J=7.2Hz, H-17α), 3.34 (1H, d, J=7.2Hz, H-17β), 1.99 (3H, s, OAc), 1.88 (1H, dt, J=12.9, 3.0Hz, H-12β), 1.45 (1H, dd, J=11.7, 4.7Hz, H-9), 1.38 (3H, s, Me-16), 1.00 (1H, dd, J= 15.6, 7.9 Hz, H-5), 0.95 (3H, s, Me-20), 0.93 (3H, s, Me-18), 0.87 (3H, s, Me-19); 13C-NMR: 43.8 (C-1), 68.4 (C-2), 46.5 (C-3), 34.6 (C-4), 55.1 (C-5), 19.5 (C-6), 35.7 (C-7), 82.3 (C-8), 53.2 (C-9), 38.9 (C-10), 17.5 (C-11), 35.9 (C-12), 106.1 (C-13), 24.1 (C-16), 73.4 (C-17), 33.6 (C-18), 22.4 (C-19), 15.5 (C-20), 170.5 (CH3CO), 21.4 (CH3CO); IR: 2945, 1737, 1455, 1393, 1250, 1025; MS: 336 ([M]+ <1%), 306 (6.1%), 215 (13.9%), 187 (41.5%), 173 (33.9%), 134 (27.3%), 107 (24.2%), 55 (19.4%), 43 (100%).
Oxidative degradation of a 1a-2a mixture with the RuCl3-NaIO4 system
To a solution of 1a-2a mixture (5 g, 14.45 mmol) in 3:4 CH3CN/H2O (70 mL) RuCl3·3H2O (50 mg) was added and the mixture stirred at room temperature. After 15 min. finely divided NaIO4 (3.1 g, 14.5 mmol) was added and the mixture was then stirred at 40°C for 5 h. The mixture was filtered and the solvent was removed down to a volume to approximately 40 mL, extracted with EtOAc (3 x 30 mL) and the combined organic layers washed successively with water, dried over Na2SO4, filtered, evaporated and chromatographed eluting with mixtures of petroleum ether/EtOAc of increasing polarity (19.8:0.2→6.4:14.6) to give 3.53 g (76%) of 5 and 0.293 g (6.3%) of 6.
Oxidative degradation of a 3-4 mixture with the KMnO4-MgSO4 system: synthesis of 2β-acetoxy-14, 15-dinor-ent-labd-8(17)-en-13-one (7)
To a solution of 3-4 mixture (2.28 g, 6.54 mmol) in acetone (50 mL), finely divided KMnO4 (1.03 g, 6.54 mmol) and MgSO4 (0.785 g, 6.54 mmol) were added in small portions during 30 minutes and the mixture stirred at room temperature. After 72 hours, the mixture was filtered, evaporated and chromatographed with petroleum ether/EtOAc mixtures of increasing polarity (19.8:0.2→15:5) yielding 1.32 g (63 %) of compound 7, 0.181 g (8.6%) of compound 5, and 0.025 g (1,2%) of compound 6. Compound 7: viscous oil; [α]D23 = -17.6° (c 9.5, CHCl3); 1H-NMR: 4.94 (1H, ddd, J=11.8, 11.8 and 4.1 Hz, H-2), 4.79 (1H, s, H-17b), 4.40 (1H, s, H-17a), 2.51 (1H, ddd, J=17.7, 8.8 and 4.6Hz, H-12b), 2.32 (1H, ddd, J=12.9, 3.9 and 2.3Hz, H-7α), 2.03 (3H, s, Me-16), 1.95 (3H, s, OAc), 1.88 (1H, ddd, J=12.9, 12.9 and 4.9Hz, H-7β), 1.57 (1H, brd, J=11.6Hz, H-9), 1.99 (1H, m, H-12a), 1.72 (1H, m, H-3α), 1.69 (1H, m, H-6β), 0.86 (3H, s, Me-18), 0.81 (3H, s, Me-19), 0.70 (3H, s, Me-20); 13C-NMR: 43.8 (C-1), 69.0 (C-2), 46.6 (C-3), 34.7 (C-4), 54.7 (C-5), 23.7 (C-6), 37.7 (C-7), 147.0 (C-8), 55.8 (C-9), 40.8 (C-10), 17.5 (C-11), 42.4 (C-12), 208.8 (C-13), 29.9 (C-16), 107.2 (C-17), 33.4 (C-18), 22.3 (C-19), 14.8 (C-20), 170.3 (CH3CO), 21.3 (CH3CO); IR: 3062, 2935, 1731, 1715, 1639, 1434, 1363, 1245, 1158, 1020; MS: 320 ([M]+ <1%), 305 (5.5%), 259 (9.7%), 245 (24.8%), 187 (42.4%), 135 (90.9%), 121 (83.0%), 119 (80.9%), 107 (73.9%), 93 (71.5%), 79 (60.1%), 43 (100%).

 ,
, {kind=link}
{kind=link}
{kind=link}


