Abstract
Cholesterylbutyrate (Chol-but) was chosen as a prodrug of butyric acid. Butyrate is not often used in vivo because its half-life is very short and therefore too large amounts of the drug would be necessary for its efficacy. In the last few years butyric acid's anti-inflammatory properties and its inhibitory activity towards histone deacetylases have been widely studied, mainly in vitro. Solid Lipid Nanoparticles (SLNs), whose lipid matrix is Chol-but, were prepared to evaluate the delivery system of Chol-but as a prodrug and to test its efficacy in vitro and in vivo. Chol-but SLNs were prepared using the microemulsion method; their average diameter is on the order of 100-150 nm and their shape is spherical. The antineoplastic effects of Chol-but SLNs were assessed in vitro on different cancer cell lines and in vivo on a rat intracerebral glioma model. The anti-inflammatory activity was evaluated on adhesion of polymorphonuclear cells to vascular endothelial cells. In the review we will present data on Chol-but SLNs in vitro and in vivo experiments, discussing the possible utilisation of nanoparticles for the delivery of prodrugs for neoplastic and chronic inflammatory diseases.
1. Introduction
Different colloidal drug delivery systems (including liposomes, dendrimers and polymeric nanoparticles, in addition to Solid Lipid Nanoparticles [SLNs]), were developed to overcome physico-chemical limitations of potential therapeutical compounds such as poor solubility, low permeability, short half-life, high molecular weight, side effects and systemic toxicity [1,2]. Butyric acid, a physiologic compound normally present in all mammalian organisms, could be regarded as a prototype of an effective in vitro anti-inflammatory and anti-cancer drug whose clinical use is heavily limited by its poor pharmacokinetic profile [3,4,5,6,7,8,9,10,11].
Cholesterylbutyrate SLNs (Chol-but SLNs), recently prepared and tested in vitro, proved to be an effective and suitable prodrug of butyrate [11,12,13,14,15]. The aims of this review are first to describe the characteristics of SLNs prepared from warm microemulsions, subsequently to analyze the biological properties of butyrate as regards to anti-inflammatory and especially anti-neoplastic, finally to discuss Chol-but SLNs in vitro and in vivo studies and their potential and future applications.
2. Theoretical and Experimental Background
2.1 Solid Lipid Nanoparticles
2.1.1 SLNs from warm microemulsions
SLNs are prepared from warm microemulsions. Microemulsions are chemical-physical systems composed of oil, water, cosurfactant and surfactant, and have an interfacial tension near zero, thus accounting for their long-term stability. The mean diameter of the microemulsion nanodroplets is below 80 nm.
Warm microemulsions are prepared at temperature ranging from 60 to 80 degrees by using melted lipids (such as fatty acids/triglycerides) and are subsequently dispersed in cold water. Nanodroplets obtained using this procedure become SLNs, which are successively washed by tangential flow filtration. SLNs are spherical in shape and with a narrow size distribution. The zeta potential is always high (30/40 mV), being negative or positive depending on the starting formulation.
Hydrophilic and lipophilic molecules (drugs or diagnostic compounds) can be incorporated in SLNs using different methods [16]. The mean diameter of drug loaded SLNs ranges from 80 to 200 nm, according to the chemical characteristics and the amount of the incorporated molecules. Drugs of different structure and lipophilicity, such as cyclosporine A, paclitaxel, doxorubicin, tobramycin, steroids, peptides, antisense oligonucleotides, or diagnostic compounds, such as Gadolinium derivatives or iron oxide, were loaded into SLNs.
2.1.2 Some peculiarities of SLNs
Internalization into cell lines: unloaded SLNs are quickly (2-5 min) uptaken by neoplastic cells in vitro. SLNs loaded with antineoplastic drugs, such as paclitaxel and doxorubicin, are also uptaken by different neoplastic cell lines. Their effect is up to 100 times higher than that observed with the drug commercial formulations and it is probably related to the fast and large internalization of SLNs into cells [17,18,19,20].
Targeting to lymph: drug-unloaded and drug-loaded SLNs, after administration in rats by duodenal/oral route, are targeted to lymph [21].
Stealth SLNs: stealth SLNs can be prepared in order to enable them to avoid their recognition by the reticulo-endothelial system (RES) after intravenous (i.v.) administration in rats, thus prolonging their residence time [22,23].
Crossing the blood-brain barrier: unloaded stealth and non-stealth SLNs, i.v. administered to rats are able to overcome the blood-brain barrier (BBB). Stealth and non-stealth SLNs loaded with different drugs reach the brain whereas free drugs are not able to efficaciously pass through the BBB [20,23,24].
2.1.3 Administration routes of SLNs
Different administration routes were tested in laboratory animals and precisely duodenal/oral, i.v., and ocular topical (eye drops) [25,26]. Melatonin-SLNs were administered in humans by oral and transdermal routes [27].
Duodenal/oral route: Tobramycin is currently administered by parenteral route. This drug was chosen to demonstrate that Tobramycin-loaded SLNs (Tobra-SLNs) are able to pass through the lymph to plasma and to evaluate Tobra-SLNs pharmacokinetics. Tobra-SLNs, injected to rats by duodenal route, easily reach plasma. Therefore, their pharmacokinetic parameters are deeply enhanced in comparison to drug-free solutions i.v. administration. So a drug, usually not absorbed by gastro-enteric route and necessarily i.v. administered, could be found in larger amounts in plasma if vehiculated by SLNs [26]. Furthermore the targeting of Tobra-SLNs to lymph is also confirmed; TEM analysis shows that even more than twenty hours after duodenal administration, Tobra-SLNs are still present in the mesenteric lymph nodes.
Intravenous administration: Non-stealth and stealth Doxorubicin-loaded SLNs (Doxo-SLNs) were prepared, the latter containing increasing amounts of stealth agent, and were i.v. administered to rabbits in order to compare their pharmacokinetic in comparison with doxorubicin free solution. Stealth Doxo-SLNs reach the brain in larger amounts whereas only a smaller proportion of non-stealth Doxo-SLNs does so; doxorubicin free solution is not able to reach the brain. These data confirm that the brain drug concentration directly depends on the amount of stealth agent used [26].
Transdermal route: SLNs loaded with melatonin, a natural hormone, were delivered by oral route and via transdermal patches in healthy volunteers with very promising clinical results [27].
In conclusion, SLNs are a sustained-release drug deliver system for several administration routes. They could also be regarded as formulations suitable to delivery by duodenal/oral administration drugs with low solubility and not absorbed by gastro-enteric tract, so avoiding parenteral route.
2.2 Butyrate
Butyric acid belongs to the family of short-chain fatty acids (SCFA), that includes also acetic, propionic and valeric acid. These compounds are physiologic metabolites present in all mammalian organisms and are largely produced in the colon by bacterial anaerobic fermentation of undigested carbohydrates (mainly derived from soluble fibres) and proteins [5,8,9,10,28,29,30]. SCFA, whose concentration is regulated by both local production-absorption rates and gut elimination, are metabolized by epithelial cells in the colon, where they act as primary energy source and play a significant role in maintaining physiologic functions [10,11,29,31,32,33,34]. Butyric acid, the most effective agent among SCFA, was evaluated as a therapeutical agent in some intestinal dysfunctions, mainly because of its anti-inflammatory and anti-cancer properties [9,10,30].
2.2.1 Anti-inflammatory properties.
Butyrate plays its crucial physiological role in maintaining the health and the integrity of the intestinal mucosa by regulating the balance between epithelial cell proliferation, differentiation and apoptosis [35]. Apart from its well known growth-preventing and apoptosis-inducing roles in neoplastic colonocytes, butyrate is able to reduce inflammation in experimental colitis and to exert in vitro trophic and anti-inflammatory effects in normal colonocytes.
The chronic inflammation that is the hallmark of Inflammatory Bowel Diseases (IBD) results from the recruitment and activation of immune cells from the circulation. These in turn release locally in the submucosa pro-inflammatory cytokines, including members of the Interleukin (IL) family and Tumour Necrosing Factor (TNF)-alpha, which play an important role in the pathogenesis of IBD [36,37]. The exact mechanism of action of butyrate on inflammation is only partially understood; however several possible mechanisms were evaluated.
In an animal model, colitis was induced in rats by oral dextran sulphate sodium (DSS) and the animals were treated either with sodium butyrate (Na-but) or saline enemas. Butyrate shows a significant protection against the decrease in cell viability, the increase in mucosal permeability, and the polymorphonuclear (PMN) cells infiltration, seen in DSS colitis. Butyrate inhibits inducible heat shock protein 70 expression in DSS colitis and the activation of heat shock factors and inducible nuclear transcriptional activator NF-κB (NF-κB) [38]. Similar data were confirmed in rat experimental colitis induced by trinitrobenzene acid: topical administration of Na-but improves the symptoms of colitis and promotes rapid repair of the epithelium in the active phase, with a reduction of IL-1β and NF-κB production [39].
In humans several clinical trials showed that oral or topical administration of butyrate induces clinical improvement/remission of Crohn’s disease and distal ulcerative colitis. Immunopathology analysis of intestinal biopsies indicates that the IL-1β secretion is significantly decreased, and a favourable downward trend is observed for IL-6 and IL-12 levels [40,41,42].
In isolated lamina propria and peripheral mononuclear cells cultured from intestinal biopsy specimen of Crohn’s patients, butyrate decreases TNF production and pro-inflammatory cytokine mRNA expression. Furthermore butyrate abolishes lipopolysaccharide induced expression of cytokine by peripheral blood mononuclear cells and transmigration of NF-κB from the cytoplasm to the nucleus [43].
Several in vitro studies confirmed the anti-inflammatory effects of butyrate using different models. Using cultured mesenteric lymph node lymphocytes it was shown that Na-but inhibits lymphocyte proliferation, IL-2 production, and Interferon (INF)-γ production [44]. These data are confirmed using the whole-blood model from healthy human volunteers: butyrate decreases TNF-α, INF-γ, IL-5, IL-12 production in peripheral blood mononuclear cells stimulated with lipopolysaccharides, whereas it does not affect the release of IL-6 [45]. Similar results are observed using human neutrophils from human blood of healthy volunteers, confirming that the involved mechanism is the suppression of NF-κB reported activity, immuno-related gene expression and cytokine release [46].
Adhesion and migration of leucocytes into the surrounding tissues is a crucial step in inflammation; a recent research investigated the effect of butyrate on the expression of endothelial leucocyte adhesion molecules by cytokine-stimulated human umbilical vein endothelial cells (HUVEC). Pre-treatment of HUVEC with butyrate inhibits TNF-α-induced expression of vascular cell adhesion molecule-1 (VCAM-1) and intracellular cell adhesion molecule (ICAM-1) in a time and concentration-dependent manner. These findings indicate that anti-inflammatory action of butyrate is partly attributable to an inhibitory expression of VCAM-1 and ICAM-1 [47,48].
2.2.2 Anti-cancer effects.
Several in vitro studies showed that butyrate acts as an anti-cancer agent, inhibiting proliferation, stimulating differentiation and inducing apoptosis in a wide panel of neoplastic cell lines (not only restricted to colorectal district but also including breast, gastric, lung, brain, pancreas) [9,10,11,19].
Butyrate is regarded as an endogenous member of the family of histone deacetylases (HDAC) inhibitors (HDACI). Dynamic acetylation-deacetylation of core histones (at the ε-amino group of lysine residues located in their N-terminal tail) is one of the various mechanisms that regulate gene expression at post-translational level [49,50]. Altered expression of HDAC as well as disequilibrium in the histone acetyltransferases (HAT)-HDAC balance are involved in the development and progression of cancer and therefore HDAC are regarded as a potential effective anticancer agents [49,50,51].
Butyrate causes a non-competitive and reversible inhibition of Class I (type 1, 2, 3, and 8) and – to a lesser degree – Class IIa (type 4, 5, 7, and 9) HDAC [51,52,53], even though more recently a competitive interaction was suggested [53]. Class I and II HDAC have a zinc pocket catalytic domain, crucial for the binding and indispensable for the inhibitory activity of HDACI [9,30,50,51,54,55,56,57]. Moreover, HDACI show varying inhibitory efficiency and specificity on different HDAC types [51] and could be classified in different ways, according to their structure and/or function [30,49,50,58,59,60]. Recently it has been suggested that SCFA (such as phenylbutyrate and valproate), due to the shortness of their side chain, could come into contact with the zinc ion but are not able to establish a significantly strong interaction with the HDAC catalytic pocket, thereby accounting for their low potency and low specificity [51,61].
Antineoplastic effects of HDACI are mediated by their ability to increase acetylation of both nuclear histones and non-histone proteins (such as transcription factors, nuclear import factors, and chaperone and cytoskeleton proteins), so inducing transcriptional and non-transcriptional effects that result in gene expression modulation and activation-inhibition of different pathways [49,50,62,63]. Furthermore, HDACI anti-cancer activities are not only limited to neoplastic cells (acting on apoptosis, cell cycle regulation, and differentiation) but also involve the regulation of host immune responses and tumour angiogenesis [51,52].
However, different mechanisms, including mRNA stabilisation and direct action on gene transcription, could cooperate to produce HDACI (and in particular Na-but) effects, as recently confirmed by genomic and proteomic studies [10,64].
Butyrate induces cell cycle arrest and proliferation prevention, facilitating or inhibiting the transcription of different regulatory genes [50]. It could promote cyclin-dependent kinase (CDK) inhibitory proteins (such as p21WAF1/CIP1 and p27KIP1) and decrease cell cycle regulatory proteins (such as cyclin D1/CDK4, CDK6, and cyclin E/CDK2) expression, so causing G0/G1 cell cycle arrest [11,30,65,66,67,68,69,70]. Furthermore, butyrate blocks G2 progression, probably either inhibiting other CDK (such as A and B1) or exploiting the lack of a G2 checkpoint in some cancer cells, as suggested for other HDACI [50,71,72,73]. Moreover, in Jurkat human T lymphocytes butyric acid influences cell cycle regulation and induces apoptosis in a dose-dependent manner, firstly by reducing the expression of G1/S regulators (such as Cyclin A, Cyclin E, CDK2, CDK4, and CDK6) and subsequently by modulating G2/M regulators (decreasing cyclin B, Cdc25c, and p27KIP1 and increasing p21CIP1/WAF1 expression) [74].
Butyric acid's pro-differentiating and anti-proliferative effects on different cell lines (such as melanoma, renal, and retinoblastoma cell lines) are dose- and time-dependent, obtaining significant activity only when the drug is delivered for more than 1 day at high concentrations (0.1-1 mM) [13,75,76,77,78,79,80].
Anti-apoptotic effects of butyrate are mediated by both p53-dependent (increase of p53 activity) and independent pathways (activation of Death Receptors 5 [DR5], TNF-related Apoptosis-inducing ligand [TRAIL], Fas, FasL and induction of expression of pro-apoptotic proteins Bax, Bak and Bik and decrease of bcl-2) [11,13,30,65,81,82,83,84,85,86,87,88,89,90].
Butyrate is also able to modulate the expression of some oncogenes (such as c-myc, c-fos, c-ras) [11,13,91,92,93], to induce tumour cell sensitization to Fas-mediated cytotoxicity [13,30,94,95], to increase susceptibility to TNF-α and IFN-γ [30,86], to decrease metastatic ability of cancer cells (i.e. inhibiting urokinase plasminogen activator [uPA] expression and inducing metastasis suppressors such as Tissue Inhibitor Matrix Metalloproteinases) [10,96].
Butyrate interferes with the inflammation-carcinogenesis pathways. For instance in colon cancer cells it reduces Cox-2 over-expression and restores Transforming Growth Factor (TGF)-β responsiveness by normalizing transcriptional rate of TGF-β receptor type I [30,97,98,99]. Furthermore, it is able to increase immunosurveillance, i.e. in rat colon cancer cells (acting on the expression of major histocompatibility complex class I and intercellular adhesion molecule 1) and in HUVEC (stimulating cell surface ICAM-1 and E-selectin expression) [8,100,101,102].
Na-but modulates the expression of genes involved in protein processing, secretion, and redox activity, therefore suggesting a possible action on ubiquitin-proteasome pathway [10,103], and reduces Vascular Endothelial Growth Factor (VEGF) expression in different human glioblastoma cell lines (T98G, U251MG, and U87MG) [104], confirming that the HDACI antineoplastic effect could be also mediated by their anti-angiogenic activity [105,106,107,108,109].
Furthermore, Na-but increases the response to radiation of cancer cells (human colon carcinoma cell lines) [110,111,112]; other HDACI share the same effect both in vitro (including glioma malignant cell lines) and in vivo (subcutaneous mice glioma xenografts) [110,113,114], probably inducing aberrant mitoses and increasing DNA double-strand breaks [114].
Gupta et al. recently outlined that the tumour suppressor gene SLC5A8 was identified as coding for a Na+-coupled plasma membrane transporter for short-chain monocarboxylates (SMCT1), including butyrate [30]. This gene is often silenced not only in colorectal but also in stomach, thyroid, prostate, breast, and brain tumours [30,115,116]. This discovery could open new research strategies to better understand the role of butyrate as anticancer agent not only within the colon but also in different tissues that share the same membrane transporter [30].
In conclusion, in vitro studies showed that butyrate displays a broad and diversified spectrum of antineoplastic actions, in some cases slightly different from other HDACI, suggesting a possible use of this drug as an effective alternative and/or synergic chemotherapic agent.
Despite these encouraging results, in vivo studies on butyrate were disappointing and did not show significant antineoplastic activities in both animal models and clinical trials [3,13], mainly because of pharmacokinetic peculiarities of this drug [3,4,5,6,7,9,10,11]. Butyrate plasma clearance is very rapid: when given intravenously its half-life is about 6 minutes [3,5,7,10]. Trials performed using large amount of butyric acid sodium or arginine salts (the latter employed to avoid toxic effects of sodium loading), given continuously or with multiple daily doses i.v. or by intraperitoneal (i.p.) infusion, clearly show that blood levels achieved are largely under the minimal concentration required to produce the same pharmacodynamic effects obtained in vitro [3,5,7,8,10,11]. Moreover butyrate first pass metabolism in liver is another limiting factor for the use of this drug in clinical practice, because of the need of strict controlled parenteral dosing [4,6,9]. Furthermore adverse events, such as anaemia, headache, nausea, diarrhoea, abdominal cramps, and strong odour, further reduce patient compliance [15,117,118].
To overcome these negative pharmacokinetic characteristics, stable derivatives or prodrugs of butyrate were studied, some of them in phase I and II clinical trials [15,119,120]. In addition these prodrugs, compared to butyrate, display more favourable peculiarities, such as better ability to cross cell membranes (including BBB), very low systemic toxicity, better oral bioavailability, and they show synergic activities if used in combination with chemotherapics and ionizing radiation [114,121,122,123,124,125,126,127].
3. Cholesterylbutyrate solid lipid nanoparticles
Chol-but was chosen as another matrix to prepare – always from warm microemulsions – Chol-but SLNs that in turn could act as a suitable and effective prodrug to deliver butyric acid, so avoiding the aforementioned negative pharmacokinetic properties and side effects of this drug.
Chol-but is the ester of cholesterol and butyric acid, two natural and physiological molecules having different functional roles in maintaining normal homeostasis in all mammalians.
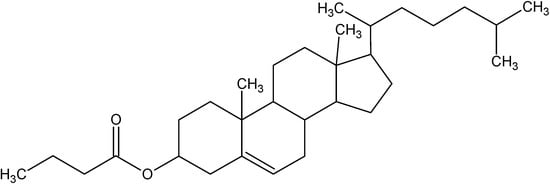
Figure 1.
Structure of Cholesterylbutyrate.
Figure 1.
Structure of Cholesterylbutyrate.
Chol-but SLNs are prepared [11,12,15,21] using the microemulsion method. Epikuron 200 (containing about 95% of soy phosphatidylcholine) (15%) and Chol-but (12%) are melted at 85°C and a warm water solution (59%) of sodium taurocholate (3%) and butanol (11%) is added to obtain a clear system. The microemulsion is immediately dispersed 1:10 in cold water and the resulting Chol-but SLNs aqueous dispersion is washed twice with water by diaultrafiltration system and successively sterilized by autoclaving (15 min at 121°C, 1 atm) before use.
Chol-but SLNs employed in the following reported studies show a mean diameter varying from 70 to 200 nm. Their polydispersity index ranges from 0.2 to 0.65 and Zeta potential is about -28 / -29 mV. Chol-but concentration of sterilized SLNs dispersion – quantified using high pressure liquid chromatography (HPLC) – varies in the different formulations from 1.7 to 30 mM.
3.1 Anti-inflammatory effects
Colloidal delivery systems, such as nanoparticles, can be designed to control drug release after oral administration and seem to be promising primarily to reduce the dosage frequency. In the case of colitis, a strong immune response is known from the inflamed regions. It was reported that drug delivery systems such as micropheres and nanoparticles can be effectively targeted to immuno-regulating cells in experimental IBD [128,129]. Thus it may be expected that either the uptake of micro- and nano-particles by the immuno-related cells or the disruption of the intestinal barrier function could allow the accumulation of the particulate carrier system in the desired area. Therefore the vehiculated drug could concentrate at the site of action, so reducing possible adverse effects and enhancing the activity of the administered dose [129]. SLNs consist of non-toxic bio-acceptable and bio-degradable substances, i.e. solid lipids and phosphatidylcholine, and their rapid internalization is probably due to their size and composition.
In a recent research we compared the effect of Na-but with Chol-but SLNs on PMN cell function, by assessing their capacity to modulate adhesion to endothelial cells (HUVEC), superoxide anion (O2-.) production, and myeloperoxidase release in vitro [15]. Our results show that Chol-but SLNs exert a striking inhibitory effect on activated PMNs, evaluated in terms of inhibition of adhesiveness, O2-. production and myeloperoxidase release, and it is much more efficient than Na-but. This improvement may be ascribed to the rapid uptake of Chol-but SLNs, whose cytoplasmic levels are already maximal after 10 min in both PMN and HUVEC. The higher inhibitory activity of adhesiveness of Chol-but SLNs (10-8 - 10-5 M) compared to Na-but might be ascribed to its accumulation within cells and to the release of higher concentrations of butyric acid from the internalized drug by esterase activities, confirming our previous data [11].
3.2 Antineoplastic effects
Antineoplastic effects of Chol-but SLNs compared to Na-but were studied in vitro on several cancer cell lines [11,12,13,14]. Pellizzaro et al. studied the anti-proliferative effects of different Chol-but SLNs compared to free Na-but in non-small-cell lung carcinoma (NSCL) cell line (NIH-H460) cultures [12]. After 6 days of treatment Na-but displays a dose-dependent anti-proliferative effect (IC50 = 0.2mM, complete growth inhibition at 2 mM). Chol-but SLNs clearly show a dose-dependent cell growth inhibition, unexpectedly inversely related to the butyric acid content. The increase in Chol-but from 1.7 to 30 mM (and the consequent increment of Chol-but/phosphatidylcholine ratio) corresponds to an increase of IC50 from 0.14 to 0.8 mM. This effect could be explained by the decreased content in phosphatidylcholine, a compound that facilitates SLNs cellular internalization. The authors found that Chol-but SLNs are able to induce 90% cell growth inhibition (IC90) at 0.19 mM (six times lower than that needed by Na-but = 1.2 mM) and complete inhibition at 0.25 mM (concentration at which Na-but causes only about 55% growth inhibition) [12,14]. Using Chol-but SLNs tagged with 6-coumarin (a green laser dye), the authors showed that these SLNs are almost completely internalized by the cells in 5 minutes [12].
In a subsequent study performed in the same in vitro model, Ugazio et al. evaluated the influence on NIH-H460 proliferation of two factors crucial in the preparation of Chol-but SLNs: microemulsion formulation (i.e. Chol-but percentage) and microemulsion/water ratio [14]. They found that, among different experimental settings, the best Chol-but SLNs formulation (0.125 mM of Chol-but – corresponding to 6% of Chol-but – and ratio = 1:80) causes complete cell growth inhibition while the same Na-but concentration provides a lower inhibition rate (only 38%).
Salomone et al. studied the anti-proliferative and pro-apoptotic activity of Chol-but SLNs at different concentrations (0.25, 0.5, and 1 mM) and incubation times on two melanoma cell lines (human MELTO1 and mouse B16) compared to free Na-but at the same concentrations [13]. The authors showed that Chol-but SLNs exert anti-proliferative and pro-apoptotic effects at lower doses and shorter treatment times than Na-but. Chol-but SLNs anti-proliferative activity is time- and dose-dependent in the first 24 hours, whereas it is strictly dose-dependent in prolonged treatments (significant reduction in cell viability after 12 hours at 0.25 mM and after only 3 hours at 0.5 and 1 mM Chol-but SLNs). Na-but induces a significant decrease in cell viability only for 0.5 and 1 mM concentrations and only after 24 hours of treatment. Similarly Chol-but SLNs induce apoptosis (using annexin V/propidium iodide staining as well bcl-2 and Fas/APO1 expression analyses) at lower concentrations and earlier than Na-but. This effect of Chol-but SLNs is dose- and time-dependent from 3 to 24 hours of treatment and only dose-dependent after 24 hours. Chol-but SLNs action on cell cycle regulation at 24 hours of treatment is characterized by significant decrease of proliferating cells (S and G2-M phases) and an increase of cells blocked in the G0/G1 to S transition phase. Cellular 6-coumarin-tagged-Chol-but SLNs uptake is almost complete after 30 minutes.
Serpe et al. studied the effects of Chol-but SLNs compared to Na-but at different concentrations (0.25 mM, 0.5 mM, and 1 mM) on three human leukemic cell lines (Jurkat from acute T cell leukemia, U937 from histiocytic lymphoma, HL-60 from promyelocytic leukemia), evaluating in particular cell-cycle distribution and c-myc expression [11]. The authors found that Chol-but SLNs are able to induce a greater cell growth inhibition (in both myeloid and lymphoid cell lines tested) than Na-but. Chol-but SLNs cause a cell-cycle arrest in G1 phase in myeloid model (U937 and HL-60) whereas mainly in G2 phase in lymphoid cells (Jurkat). If the block in G0/G1 phase is mainly ascribed to c-myc repression and/or to p21WAF1 up-regulation, the block in G2/M phase is unexpected. However these data are in agreement with the previously reported results of the work by Kurita-Ochiai et al. [74]. C-myc expression is rapidly and transiently down-regulated in all the three cell lines after treatment with Chol-but SLNs 0.25mM whilst it is slightly decreased only in U937 cells after treatment with Na-but at higher concentrations (1 mM).
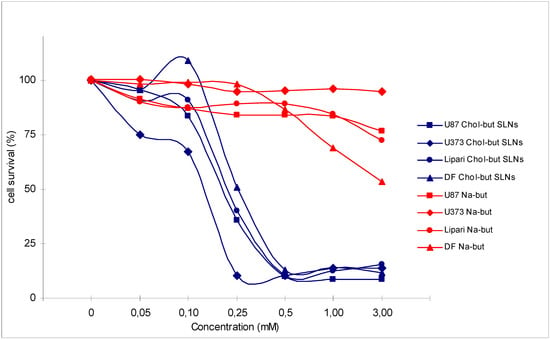
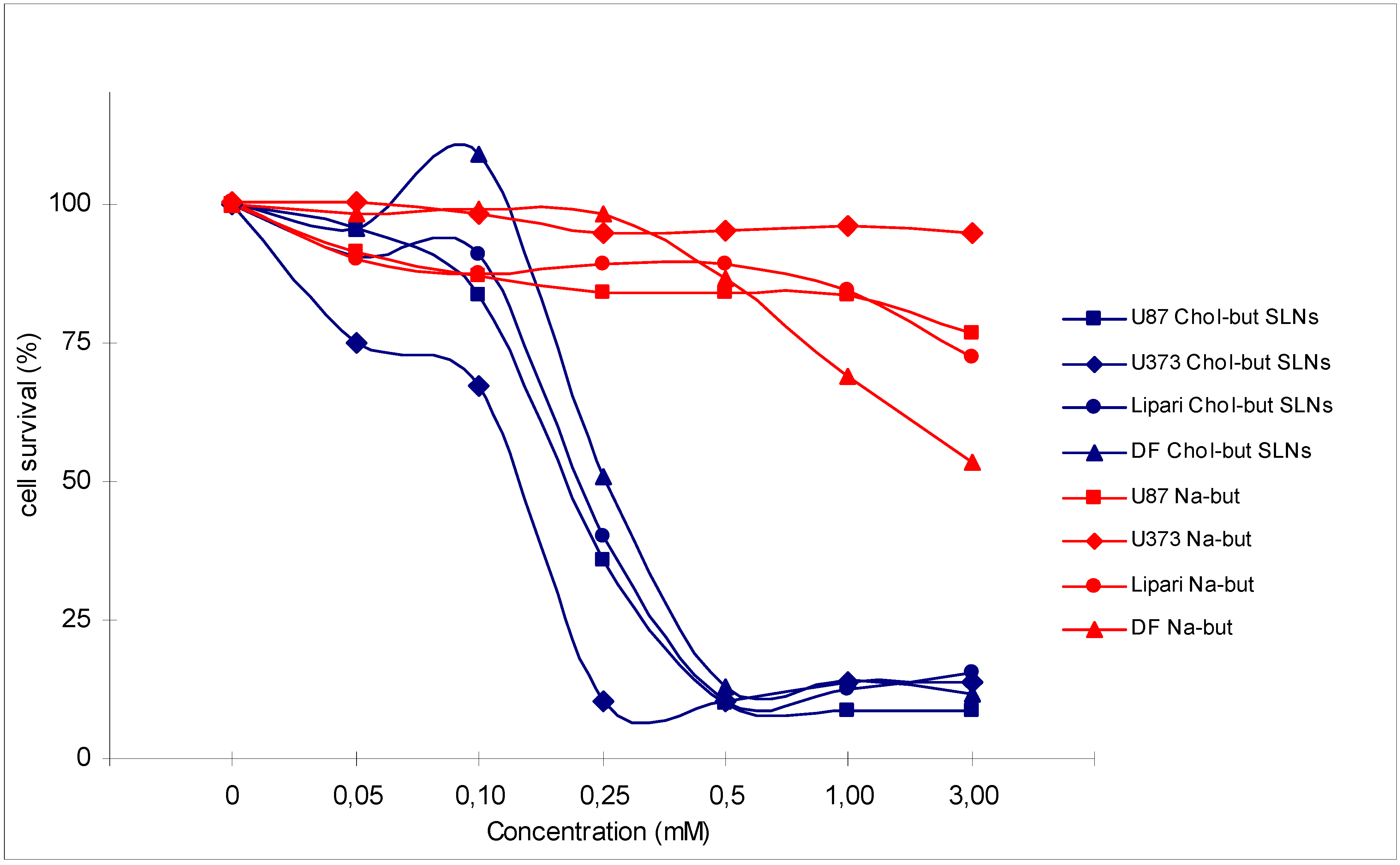
Figure 2.
Cytotoxic activity analysis (MTT test) performed on 4 human glioma cell cultures (U87, U373, Lipari, DF) treated with Na-but and Chol-but SLNs at different concentrations after 72 hours.
Figure 2.
Cytotoxic activity analysis (MTT test) performed on 4 human glioma cell cultures (U87, U373, Lipari, DF) treated with Na-but and Chol-but SLNs at different concentrations after 72 hours.
Chol-but SLNs induce much more apoptotic cell death compared to Na-but at the same concentrations. Flow cytometry DNA analysis and annexin V labelling clearly show marked increase in the proportion of cells with DNA fragmentation and blocked in G2/M phase compared to Na-but. More in details, flow cytometry DNA analysis, performed on U373 human glioma cell cultures treated with Chol-but SLNs 0.125 mM, shows a significant amount of apoptotic cells (20%) after 24 hours and a further increase after 48 hours (29.5%). From the earliest to latest times (24 up to 48 hours) a similar increase in the proportion of G2/M phase blocked cells and in iperdiploid DNA total content is detectable.
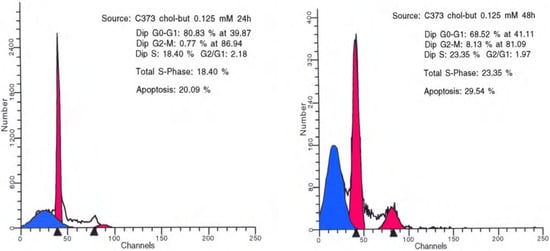
Figure 3.
Flow cytometry DNA analysis performed on U373 human glioma cell cultures treated with Chol-but SLNs 0.125 mM after 24 (left) and 48 (right) hours.
Figure 3.
Flow cytometry DNA analysis performed on U373 human glioma cell cultures treated with Chol-but SLNs 0.125 mM after 24 (left) and 48 (right) hours.
These data show that Chol-but SLNs are able to induce significantly higher anti-proliferative and pro-apoptotic effect in different glioma cell cultures compared to Na-but. Nevertheless in our in vitro model Chol-but SLNs do not produce significant pro-differentiating action, as proved by the lack of an increase in Glial Fibrillary Acidic Protein (GFAP) expression in immunohistochemical studies.
In order to confirm that Chol-but SLNs effects are not related to cholesterol or to other SLNs component, we treated the same glioma cell lines with Palmitate-SLNs (containing Palmitate instead of Chol-but) at the same concentrations. Palmitate was chosen because of its well previously demonstrated ineffectiveness in different cell cultures. Treatment with Palmitate-SLNs does not produce neither anti-proliferative nor pro-apoptotic changes in all the glioma cell lines.
In vivo Wistar rats bearing intracerebral stereotactic C6 cell implants (1 x 105 cells injected into right caudate nucleus) were treated with Chol-but SLNs 30 mg/kg (corresponding to a concentration of 29 mM of sterilized SLNs dispersion) daily i.v. infusion for 7 consecutive days (treated animals) or with saline (control animals). One day randomly chosen among the seven days of treatment, Fluorescent Chol-but SLNs (containing 6-coumarin 0.04%) instead of Chol-but SLNs were administered in different groups of animals. On day 21st brain tumour samples were obtained for morphological and immunohistochemical analyses. A significant shrinkage in tumours of treated animals is evident. Residual tumour cells (maintaining infiltrating behaviour and nodular aspects similar to that displayed by not treated tumours) are located at the border of the implanted area, which is replaced by cellular debris or is completely empty. Most of these cancer cells display morphologic and immunohistochemical apoptotic features. Immunofluorescence analyses clearly show that Chol-but SLNs are rapidly internalized in glioma cells where persist for few days, possibly undergoing intracellular metabolism as demonstrated by different subcellular localisations observed in subsequent days.
In conclusion, in vitro studies on different cancer cell lines suggest that Chol-but SLNs are able to induce anti-proliferative and pro-apoptotic effects earlier and at significantly lower concentrations compared to Na-but. This result is confirmed in our preliminary in vivo study on rat glioma model in which Chol-but SLNs display an unexpected high effectiveness. Anti-proliferative and pro-apoptotic actions are time- and dose-dependent in the first hours of treatment (within 24 hours) and only dose-dependent for prolonged times. From these studies we could suggest that the mechanisms underlying these processes seem to follow the same pathways involved in butyrate activity but few discrepancies have to be highlighted. For instance the anti-proliferative effect appears to be mainly mediated by blockage in G0/G1 phases; however a significant increase in G2/M block compared to Na-but is observed in some (but not in all) of cancer cell lines tested. Furthermore, after Chol-but SLNs treatments the percentage of apoptotic cells seems to be higher compared to Na-but, even at later times (more than 24 hours). These results support the hypothesis that the mechanisms of action of Chol-but SLNs do not accurately overlap Na-but, so opening new therapeutical perspectives.
The pro-differentiating effect described after Na-but treatments are not confirmed for Chol-but SLNs, even if the data supporting this statement are too small.
4. Discussion – Conclusions
In this review, according to the results of in vitro and in vivo studies, we showed that Chol-but SLNs could be regarded as suitable and highly effective prodrug of butyric acid. In the meantime we emphasized that Chol-but SLNs display chemical-physical characteristics and pharmacokinetic-pharmacodynamic properties similar to other SLNs formulations. More in details, Chol-but SLNs are composed by biocompatible and safe molecules, display spherical shape and favourable zeta potential. Moreover, Chol-but SLNs are rapidly, consistently, and persistently entrapped in intracellular compartment, so allowing a possible prolonged drug release, and do not substantially modify the specific effect of the active vehiculated drug.
In conclusion, Chol-but SLNs could be considered to all intents as “common” SLNs acting themselves as butyric acid prodrug, therefore displaying the same peculiar advantages and disadvantages.
Similarly to other non selective HDACI [130] Chol-but SLNs seem to act modulating in a non-specific manner different pathways, mainly involved in cell survival and proliferation. In cancer therapy this loss of specificity, on one hand, provides advantages over other chemotherapic agents (usually targeted against only one or few pathways) but, on the other hand, it could represent a severe limitation because of undesired and unavoidable local and systemic effects.
Nevertheless, the finding that in vitro and in vivo some neoplastic cells display resistance even to more selective HDACI (such as vorinostat) requires further efforts to better understand at molecular levels the genotype-response correlation between the drug studied and the targeted tumour cell subtype(s) [131,132]. Furthermore, functional genetic approaches directed to assess in detail the pathways modulated by each HDACI could open new therapeutic strategies, identifying potential synergic concomitant or sequential treatments (chemotherapics, other drugs, gene therapy, or radiotherapy) for genotypically (and consequently phenotypically) defined cancer cell populations [131,132]. For instance co-treatment with retinoic acid shows to enhance the HDACI anti-neoplastic effectiveness [131]. Moreover Bcl-2 and pro-apoptotic BH3-only proteins Bid and Bim influence the response to vorinostat in a B cell lymphoma model, suggesting that patients addressed to receive HDACI treatment could be selected according to the expression levels (and/or functional status) of these apoptotic markers [132].
Considering the ability of SLNs to easily pass through the BBB and to reach the central nervous system (CNS) [16,20,24,26,133,134], one of the most interesting field of research and possible therapeutical application of Chol-but SLNs is the treatment of different neurological diseases.
First of all Chol-but SLNs could prove safe and effective in the treatment of primary and/or metastatic brain tumours. Likewise SLNs in vivo results and according to previously preliminary reported data on animal studies, Chol-but SLNs are able to reach the CNS and thereby the implanted tumours, and at the same time they could avoid significant systemic toxicity [16,20,24,26,133,134,135]. Nevertheless, even if at lower doses compared to the drug free formulations, SLNs could reach every region of CNS where they could produce undesired or even dangerous effects [20]. However, this presumed disadvantage could in turn offer further therapeutical strategies, for example in the case of multifocal or metastatic brain neoplasms. In any case, other in vivo studies are needed to better assess Chol-but SLNs effectiveness in brain tumours as well as their possible distant brain areas actions and systemic effects in phase I and II trials.
Better understanding of Chol-but SLNs mechanisms of action at molecular level could help to develop Chol-but SLNs carrying one or more antineoplastic agent(s) (e.g. gene constructs and chemotherapeutic drugs), in order to modulate different pathways involved in tumour progression, not only those affected by butyrate. This could lead to develop a multitargeted therapy, administered using a single vehicle (Chol-but SLNs), specifically designed in accordance with tumour molecular characteristic profile (for example EGFR, CDK4, and p53 expression status in glioblastomas) and therefore patients’ stratification [136,137,138]. Moreover, it is possible to design Chol-but SLNs able to release each vehiculated drug(s) at different rates providing concomitant or sequential treatment, possibly time related to radiotherapy. This effort could not only enhance treatment selectivity but also further on lead to reduce the total amount of Chol-but SLNs administered, therefore reducing undesired effects on healthy tissues.
Furthermore, Chol-but SLNs display a role in modifying spontaneous immune response, in particular in glioma tumours, as well as shown in vitro for Na-but [10,97,98,99,101,102,119]. Chol-but SLNs could act as a non-specific stimulating agent of an already present but weak host immune reaction [139]. Nevertheless, Chol-but SLNs could also vehiculate different specific antigens directly into tumours, so actively provoking a more targeted anti-tumoral immune response [139].
Chol-but SLNs are already a promising drug to be used in patients affected by ulcerative colitis and Crohn’s disease; they exert a striking inhibitory effect on activated PMNs, evaluated in terms of inhibition of adhesiveness, O2-. production and myeloperoxidase release, and it is much more efficient than Na-but. These suggestions are further strengthened by our recent preliminary data showing that Chol-but SLNs are able to decrease IL and TNF-α secretion from human lymphocytes.
Another field in which Chol-but SLNs could offer new therapeutical application is represented by neurodegenerative diseases.
In the recent years increasing experimental evidences showed a possible direct or indirect involvement of epigenetic modifications in neurodegenerative diseases. More in details, a deregulated balance between HDAC and HAT, resulting in impaired chromatin acetylation status, is involved in polyglutamine-related disorders (such as Huntington disease and spinocerebellar ataxia type 1), motor neuron diseases (such as spinal muscular atrophy [SMA], spinal and bulbar muscular atrophy [SBMA], and amyotrophic lateral sclerosis), ischemia, and oxidative stress induced neurodegenerative diseases (such as Alzheimer’s and Parkinson’s diseases). These data suggest a possible therapeutical role of HDACI in these disorders [140,141,142]. Until now few HDACI were studied in vitro and in vivo in order to evaluate their ability to restore normal levels of acetylation and to increase neuronal survival [141].
More in details, in several in vitro and in vivo models of Huntington disease Na-but is able to improve motor performance, to prolong survival and to ameliorate neuropathological changes (striatal atrophy) [140,143,144]. A dose-finding study of the HDACI phenylbutyrate in Huntington’s disease patients shows a significant decrease in a blood biomarker (constituted by 322 mRNAs) in treated compared to not treated subjects [145]. Conversely, encouraging results obtained in animal models of SMA [146] and SBMA [147] treated with Na-but are not confirmed in a recent randomized clinical trial in SMA patients taking phenylbutyrate [148]. Similar discordant results regarding the use of different HDACI in neurodegenerative disorders are found in other in vitro studies and could be explained by tissue specific expression of different classes of HDAC [142]. Consequently a great effort has to be made to design more brain-specific and/or selective HDACI [142].
Although Chol-but SLNs have to be regarded as non selective HDACI, their usefulness in neurodegenerative diseases – beyond their intrinsic activities – could be primarily related to their aforementioned pharmacokinetic and pharmacodynamic peculiarities. More in details, Chol-but SLNs are able to easily pass through the BBB, thus reducing the amount of the drug used and consequently decreasing both systemic and local toxicity. Moreover, Chol-but SLNs could vehiculate more than one therapeutic agent and allow prolonged and modulated drug(s) release. These properties could give some advantages to Chol-but SLNs, compared to more specific antineoplastic or immunomodulating systemically administered drugs. For instance, new experimental evidence suggests that the field of action of HDACI could also be expanded to mixed inflammatory and degenerative disorders such as multiple sclerosis [140]. In this heterogeneous disease, inflammation is regarded as the first etiopathogenetic actor and is the main target of the immunomodulatory therapies currently in use. However, inflammation could not completely justify the highly variable clinical course, especially in long lasting or in peculiar forms of this disorder, such as primary progressive, in which neuronal loss seems to predominate. This is an intriguing statement because it could represent the point of contact between anti-inflammatory and neuroprotective actions of HDACI and could open for Chol-but SLNs new therapeutical approaches in a broader spectrum of immunological and degenerative diseases.
Acknowledgements
We thank Dr. Lorenzo Priano and Dr. Matteo Bigoni for helpful critical discussion and revision of the manuscript. This paper is in memory of our beloved friends and collegues Cinzia Miscio and Simonetta D’Alisa.
References and Notes
- Gasco, M.R. Lipid nanoparticles: perspectives and challenges. Adv. Drug Deliv. Rev. 2007, 59, 377–378. [Google Scholar] [CrossRef]
- Müller, R.H. Lipid nanoparticles: recent advances. Adv. Drug Deliv. Rev. 2007, 59, 375–376. [Google Scholar] [CrossRef]
- Miller, A.A.; Kurschel, E.; Osieka, R.; Schmidt, C.G. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur. J. Cancer Clin. Oncol. 1987, 23, 1283–1287. [Google Scholar] [CrossRef]
- Perrine, S.P.; Ginder, G.D.; Faller, D.V.; Dover, G.H.; Ikuta, T.; Witkowska, H.E.; Cai, S.P.; Vichinsky, E.P.; Olivieri, N.F. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N. Engl. J. Med. 1993, 328, 81–86. [Google Scholar] [CrossRef]
- Newmark, H.L.; Young, C.W. Butyrate and phenylacetate as differentiating agents: practical problems and opportunities. J. Cell Biochem. Suppl. 1995, 22, 247–253. [Google Scholar] [CrossRef]
- Pouillart, P.R. Role of butyric acid and its derivatives in the treatment of colorectal cancer and hemoglobinopathies. Life Sci. 1998, 63, 1739–1760. [Google Scholar] [CrossRef]
- Egorin, M.J.; Yuan, Z.M.; Sentz, D.L.; Plaisance, K.; Eiseman, J.L. Plasma pharmacokinetics of butyrate after intravenous administration of sodium butyrate or oral administration of tributyrin or sodium butyrate to mice and rats. Cancer Chemother. Pharmacol. 1999, 43, 445–453. [Google Scholar] [CrossRef]
- Santini, V.; Zozzini, A.; Scappini, B.; Grossi, A.; Rossi Ferrini, P. Searching for the magic bullet against cancer: the butyrate saga. Leuk. Lymphoma. 2001, 42, 275–289. [Google Scholar] [CrossRef]
- Chen, J.S.; Faller, D.V.; Spanjaard, R.A. Short-chain fatty acid inhibitors of histone deacetylases: promising anticancer therapeutics? Curr. Cancer Drug Targets 2003, 3, 219–236. [Google Scholar] [CrossRef]
- Miller, S.J. Cellular and physiological effects of short-chain fatty acids. Mini Rev. Med. Chem. 2004, 4, 839–845. [Google Scholar] [CrossRef]
- Serpe, L.; Laurora, S.; Pizzimenti, S.; Ugazio, E.; Ponti, R.; Canapaio, R.; Briatore, F.; Barrera, G.; Gasco, M.R.; Bernengo, M.G.; Eandi, M.; Zara, G.P. Cholesteryl butyrate solid lipid nanoparticles as a butyric acid pro-drug: effects on cell proliferation, cell-cycle distribution and c-myc expression in human leukemic cells. Anticancer Drugs. 2004, 15, 525–536. [Google Scholar] [CrossRef]
- Pellizzaro, C.; Cordini, D.; Morel, S.; Ugazio, E.; Gasco, M.R.; Dandone, M.G. Cholesteryl butyrate in solid lipid nanospheres as an alternative approach for butyric acid delivery. Anticancer Res. 1999, 19, 3921–3925. [Google Scholar]
- Salomone, B.; Ponti, R.; Gasco, M.R.; Ugazio, E.; Quaglino, P.; Osella-Abate, S.; Bernengo, M.G. In vitro effects of cholesteryl butyrate solid lipid nanospheres as a butyric acid pro-drug on melanoma cells: evaluation of antiproliferative activity and apoptosis induction. Clin. Exp. Metastasis 2001, 18, 663–673. [Google Scholar]
- Ugazio, E.; Marengo, E.; Pellizzaro, C.; Coradini, D.; Peira, E.; Daidone, M.G.; Gasco, M.R. The effect of formulation and concentration of cholesteryl butyrate solid lipid nanospheres (SLN) on NIH-H460 cell proliferation. Eur. J. Pharm. Biopharm. 2001, 52, 197–202. [Google Scholar] [CrossRef]
- Dianzani, C.; Cavalli, R.; Zara, G.P.; Gallicchio, M.; Lombardi, G.; Gasco, M.R.; Panzanelli, P.; Fantozzi, R. Cholesteryl butyrate solid lipid nanoparticles inhibit adhesion of human neutrophils to endothelial cells. Br. J. Pharmacol. 2006, 148, 648–656. [Google Scholar] [CrossRef]
- Peira, E.; Marzola, P.; Podio, V.; Aime, S.; Sbarbati, A.; Gasco, M.R. In vitro and in vivo study of solid lipid nanoparticles loaded with superparamagnetic iron oxide. J. Drug Target. 2003, 11, 19–24. [Google Scholar] [CrossRef]
- Mauro, A.; Miglietta, A.; Cavalli, R.; Bocca, C.; Guido, M.; Di Sapio, A.; Pradotto, L.; Schiffer, D.; Gasco, M.R. Enhanced cytotoxycity of Paclitaxel incorporated in Solid Lipid Nanoparticles against human glioma cells. Proceed. Int’l. Symp. Control. Rel. Bioact. Mater. 2000, 27, 377–378. [Google Scholar]
- Miglietta, A.; Cavalli, R.; Bocca, C.; Gabriel, L.; Gasco, M.R. Cellular uptake and cytotoxicity of solid lipid nanospheres (SLN) incorporating doxorubicin or paclitaxel. Int. J. Pharm. 2000, 210, 61–67. [Google Scholar] [CrossRef]
- Serpe, L.; Guido, M.; Canaparo, R.; Muntoni, E.; Cavalli, R.; Panzanelli, P.; Della Pepal, C.; Bargoni, A.; Mauro, A.; Gasco, M.R.; Eandi, M.; Zara, G.P. Intracellular accumulation and cytotoxicity of doxorubicin with different pharmaceutical formulations in human cancer cell lines. J. Nanosci. Nanotechnol. 2006, 6, 3062–3069. [Google Scholar] [CrossRef]
- Brioschi, A.; Zenga, F.; Zara, G.P.; Gasco, M.R.; Ducati, A.; Mauro, A. Solid lipid nanoparticles: could they help to improve the efficacy of pharmacologic treatments for brain tumors? Neurol. Res. 2007, 29, 324–330. [Google Scholar] [CrossRef]
- Bargoni, A.; Cavalli, R.; Caputo, O.; Fundarò, A.; Gasco, M.R.; Zara, G.P. Solid lipid nanoparticles in lymph and plasma after duodenal administration to rats. Pharm. Res. 1998, 15, 745–750. [Google Scholar] [CrossRef]
- Bocca, C.; Caputo, O.; Cavalli, R.; Gabriel, L.; Miglietta, A.; Gasco, M.R. Phagocytic uptake of fluorescent stealth and non-stealth solid lipid nanoparticles. Int. J. Pharm. 1998, 175, 185–193. [Google Scholar] [CrossRef]
- Podio, V.; Zara, G.P.; Carazzonet, M.; Cavalli, R.; Gasco, M.R. Biodistribution of stealth and non-stealth solid lipid nanospheres after intravenous administration to rats. J. Pharm. Pharmacol. 2000, 52, 1057–1063. [Google Scholar] [CrossRef]
- Zara, G.P.; Cavalli, R.; Bargoni, A.; Fundarò, A.; Vighetto, D.; Gasco, M.R. Intravenous administration to rabbits of non-stealth and stealth doxorubicin-loaded solid lipid nanoparticles at increasing concentrations of stealth agent: pharmacokinetics and distribution of doxorubicin in brain and other tissues. J. Drug Target. 2002, 10, 327–335. [Google Scholar] [CrossRef]
- Cavalli, R.; Bargoni, A.; Podio, V.; Muntoni, E.; Zara, G.P.; Gasco, M.R. Duodenal administration of solid lipid nanoparticles loaded with different percentages of tobramycin. J. Pharm. Sci. 2003, 92, 1085–1094. [Google Scholar] [CrossRef]
- Zara, G.P.; Bargoni, A.; Cavalli, R.; Fundarò, A.; Vighetto, D.; Gasco, M.R. Pharmacokinetics and tissue distribution of idarubicin-loaded solid lipid nanoparticles after duodenal administration to rats. J. Pharm. Sci. 2002, 91, 1324–1333. [Google Scholar] [CrossRef]
- Priano, L.; Esposti, D.; Esposti, R.; Castagna, G.; De Medici, C.; Fraschini, F.; Gasco, M.R.; Mauro, A. Solid Lipid Nanoparticles incorporating melatonin as a new model for sustained oral and transdermal delivery systems. J. Nanosci. Nanotechnol. 2007, 7, 1–6. [Google Scholar]
- Cummings, J.H. Short chain fatty acids in the human colon. Gut 1981, 22, 763–779. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Gupta, N.; Martin, P.M.; Prasad, P.D.; Ganapathy, V. SLC5A8 (SMCT1)-mediated transport of butyrate forms the basis for the tumor suppressive function of the transporter. Life Sci. 2006, 78, 2419–2425. [Google Scholar] [CrossRef]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef]
- Kruh, J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol. Cell Biochem. 1982, 42, 65–82. [Google Scholar]
- Jaskiewicz, J.; Zhao, Y.; Hawes, J.W.; Shimomura, Y.; Crabb, D.W.; Harris, R.A. Catabolism of isobutyrate by colonocytes. Arch. Biochem. Biophys. 1996, 327, 265–270. [Google Scholar] [CrossRef]
- Bach Knudsen, K.E.; Serena, A.; Canibe, N.; Juntunen, K.S. New insight into butyrate metabolism. Proc. Nutr. Soc. 2003, 62, 81–86. [Google Scholar] [CrossRef]
- Sartor, R.B. Pathogenesis and immune mechanism of chronic inflammatory bowel diseases. Am. J. Gastroenterol. 1997, 92 (12 Suppl.), 5S–11S. [Google Scholar]
- Brynskov, J.; Nielsen, O.H.; Ahnfelt-Rønne, I.; Bendtzen, K. Cytokines (immunoinflammatory hormones) and their natural regulation in inflammatory bowel disease (Crohn’s disease and ulcerative colitis): a review. Dig. Dis. 1994, 12, 290–304. [Google Scholar] [CrossRef]
- Fuss, I.J. Cytokines network in inflammatory bowel disease. Curr. Drug Targets Inflamm. Allergy 2003, 2, 101–112. [Google Scholar] [CrossRef]
- Venkatraman, A.; Ramakrishna, B.S.; Shaji, R.V.; Kumar, N.S.; Pulimood, A.; Patra, S. Amelioration of dextran sulfate colitis by butyrate: role of heat shock protein 70 and NF-kappaB. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G177–G184. [Google Scholar]
- Song, M.; Xia, B.; Li, J. Effects of topical treatment of sodium butyrate and 5-aminosalicylic acid on expression of trefoil factor 3, interleukin 1beta, and nuclear factor kappaB in trinitrobenzene sulphonic acid induced colitis in rats. Postgrad. Med. J. 2006, 82, 130–135. [Google Scholar] [CrossRef]
- Vernia, P.; Marcheggiano, A.; Caprilli, R.; Frieri, G.; Corrao, G.; Valpiani, D.; Di Paolo, M.C.; Paoluzi, P.; Torsoli, A. Short-chain fatty acid topical treatment in distal ulcerative colitis. Aliment. Pharmacol. Ther. 1995, 9, 309–313. [Google Scholar]
- Vernia, P.; Annese, V.; Bresci, G.; d’Albasio, G.; D'Incà, R.; Giaccari, S.; Ingrosso, M.; Mansi, C.; Riegler, G.; Valpiani, D.; Caprilli, R.; Gruppo Italiano per lo Studio del Colon and del Retto. Topical butyrate improves efficacy of 5-ASA in refractory distal ulcerative colitis: results of a multicentre trial. Eur. J. Clin. Invest. 2003, 33, 244–248. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Morera, R.; Ciccocioppo, R.; Cazzola, P.; Gotti, S.; Tinozzi, F.P.; Tinozzi, S.; Corazza, G.R. Oral butyrate for mildly to moderately active Crohn's disease. Aliment. Pharmacol. Ther. 2005, 22, 789–794. [Google Scholar] [CrossRef]
- Segain, J.P.; Raingeard de la Blétière, D.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottière, H.M.; Galmiche, J.P. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef]
- Cavaglieri, C.R.; Nishiyama, A.; Fernandes, L.C.; Curi, R.; Miles, E.A.; Calder, P.C. Differential effects of short-chain fatty acids on proliferation and production of pro- and anti-inflammatory cytokines by cultured lymphocytes. Life Sci. 2003, 73, 1683–1690. [Google Scholar] [CrossRef]
- Nancey, S.; Bienvenu, J.; Coffin, B.; Andre, F.; Descos, L.; Flourié, B. Butyrate strongly inhibits in vitro stimulated release of cytokines in blood. Dig. Dis. Sci. 2002, 47, 921–928. [Google Scholar] [CrossRef]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar]
- Menzel, T.; Lührs, H.; Zirlik, S.; Schauber, J.; Kudlich, T.; Gerke, T.; Gostner, A.; Neumann, M.; Melcher, R.; Scheppach, W. Butyrate inhibits leukocyte adhesion to endothelial cells via modulation of VCAM-1. Inflamm. Bowel Dis. 2004, 10, 122–128. [Google Scholar] [CrossRef]
- Zapolska-Downar, D.; Siennicka, A.; Kaczmarczyk, M.; Kołodziej, B.; Naruszewicz, M. Butyrate inhibits cytokine-induced VCAM-1 and ICAM-1 expression in cultured endothelial cells: the role of NF-kappaB and PPARalpha. J. Nutr. Biochem. 2004, 15, 220–228. [Google Scholar] [CrossRef]
- Balakin, K.V.; Ivanenkov, Y.A.; Kiselyov, A.S.; Tkachenko, S.E. Histone deacetylase inhibitors in cancer therapy: latest developments, trends and medicinal chemistry perspective. Anticancer Agents Med. Chem. 2007, 7, 576–592. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Kelly, W.K. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J. 2007, 13, 23–29. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Bhalla, K.N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar] [CrossRef]
- Sekhavat, A.; Sun, J.M.; Davie, J.R. Competitive inhibition of histone deacetylase activity by trichostatin A and butyrate. Biochem. Cell. Biol. 2007, 85, 751–758. [Google Scholar] [CrossRef]
- Gray, S.G.; Ekström, T.J. The human histone deacetylase family. Exp. Cell Res. 2001, 262, 75–83. [Google Scholar] [CrossRef]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef]
- Zhu, W.G.; Otterson, G.A. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr. Med. Chem. Anticancer Agents 2003, 3, 187–199. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Miller, T.; Kelly, W.K. Histone deacetylase inhibitors. Adv. Cancer Res. 2004, 91, 137–168. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Entin-Meer, M.; Rephaeli, A.; Yang, X.; Nudelman, A.; VandenBerg, S.R.; Haas-Kogan, D.A. Butyric acid prodrugs are histone deacetylase inhibitors that show antineoplastic activity and radiosensitizing capacity in the treatment of malignant gliomas. Mol. Cancer Ther. 2005, 4, 1952–1961. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Jiang, Z.; Sharfstein, S.T. Sodium butyrate stimulates monoclonal antibody over-expression in CHO cells by improving gene accessibility. Biotechnol. Bioeng. 2007. [Google Scholar]
- Litvak, D.A.; Evers, B.M.; Hwang, K.O.; Hellmich, M.R.; Ko, T.C.; Townsend, C.M., Jr. Butyrate-induced differentiation of Caco-2 cells is associated with apoptosis and early induction of p21Waf1/Cip1 and p27Kip1. Surgery 1998, 124, 161–169. [Google Scholar] [CrossRef]
- Lallemand, F.; Courilleau, D.; Buquet-Fagot, C.; Atfi, A.; Montagne, M.N.; Mester, J. Sodium butyrate induces G2 arrest in the human breast cancer cells MDA-MB-231 and renders them competent for DNA rereplication. Exp. Cell Res. 1999, 247, 432–440. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 10014–10019. [Google Scholar] [CrossRef]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef]
- Kuefer, R.; Hofer, M.D.; Altug, V.; Zorn, C.; Genze, F.; Kunzi-Rapp, K.; Hautmann, R.E.; Gschwend, J.E. Sodium butyrate and tributyrin induce in vivo growth inhibition and apoptosis in human prostate cancer. Br. J. Cancer 2004, 90, 535–541. [Google Scholar] [CrossRef]
- Kim, J.; Park, H.; Im, J.Y.; Choi, W.S.; Kim, H.S. Sodium butyrate regulates androgen receptor expression and cell cycle arrest in human prostate cancer cells. Anticancer Res. 2007, 27, 3285–3292. [Google Scholar]
- el-Deiry, W.S.; Harper, J.W.; O'Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; Wiman, K.G.; Mercer, W.E.; Kastan, M.B.; Kohn, K.W.; Elledge, S.J.; Kinzler, K.W.; Vogelstein, B. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar]
- Qiu, L.; Burgess, A.; Fairlie, D.P.; Leonard, H.; Parsons, P.G.; Gabrielli, B.G. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef]
- Burgess, A.J.; Pavey, S.; Warrener, R.; Hunter, L.J.; Piva, T.J.; Musgrove, E.A.; Saunders, N.; Parsons, P.G.; Gabrielli, B.G. Up-regulation of p21(WAF1/CIP1) by histone deacetylase inhibitors reduces their cytotoxicity. Mol. Pharmacol. 2001, 60, 828–837. [Google Scholar]
- Kurita-Ochiai, T.; Hashizume, T.; Yonezawa, H.; Ochiai, K.; Yamamoto, M. Characterization of the effects of butyric acid on cell proliferation, cell cycle distribution and apoptosis. FEMS Immunol. Med. Microbiol. 2006, 47, 67–74. [Google Scholar] [CrossRef]
- Nudelman, A.; Ruse, M.; Aviram, A.; Rabizadeh, E.; Shaklai, M.; Zimrah, Y.; Rephaeli, A. Novel anticancer prodrugs of butyric acid. 2. J. Med. Chem. 1992, 35, 687–694. [Google Scholar] [CrossRef]
- Vecchia, M.G.; Carnelós Filho, M.; Fellipe, C.R.; Curi, R.; Newsholme, E.A. Acetate and propionate potentiate the antiproliferative effect of butyrate on RBL-2H3 growth. Gen. Pharmacol. 1997, 29, 725–728. [Google Scholar] [CrossRef]
- Siu, L.L.; Von Hoff, D.D.; Rephaeli, A.; Izbicka, E.; Cerna, C.; Gomez, L.; Rowinsky, E.K.; Eckhardt, S.G. Activity of pivaloyloxymethyl butyrate, a novel anticancer agent, on primary human tumor colony-forming units. Invest. New Drugs 1998, 16, 113–119. [Google Scholar] [CrossRef]
- Madigan, M.C.; Chaudhri, G.; Penfold, P.L.; Conway, R.M. Sodium butyrate modulates p53 and Bcl-2 expression in human retinoblastoma cell lines. Oncol. Res. 1999, 11, 331–337. [Google Scholar]
- Hara, I.; Miyake, H.; Hara, S.; Arakawa, S.; Kamidono, S. Sodium butyrate induces apoptosis in human renal cell carcinoma cells and synergistically enhances their sensitivity to anti-Fas-mediated cytotoxicity. Int. J. Oncol. 2000, 17, 1213–1218. [Google Scholar]
- Giermasz, A.; Makowski, M.; Kozłowska, E.; Nowis, D.; Maj, M.; Jalili, A.; Feleszko, W.; Wójcik, C.; Dabrowska, A.; Jakóbisiak, M.; Gołab, J. Potentiating antitumor effects of a combination therapy with lovastatin and butyrate in the Lewis lung carcinoma model in mice. Int. J. Cancer 2002, 97, 746–750. [Google Scholar] [CrossRef]
- Hague, A.; Manning, A.M.; Hanlon, K.A.; Huschtscha, L.I.; Hart, D.; Paraskeva, C. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int. J. Cancer 1993, 55, 498–505. [Google Scholar] [CrossRef]
- Mandal, M.; Kumar, R. Bcl-2 expression regulates sodium butyrate-induced apoptosis in human MCF-7 breast cancer cells. Cell Growth Differ. 1996, 7, 311–318. [Google Scholar]
- Janson, W.; Brandner, G.; Siegel, J. Butyrate modulates DNA-damage-induced p53 response by induction of p53-independent differentiation and apoptosis. Oncogene 1997, 15, 1395–1406. [Google Scholar]
- Fan, Y.Y.; Zhang, J.; Barhoumi, R.; Burghardt, R.C.; Turner, N.D.; Lupton, J.R.; Chapkin, R.S. Antagonism of CD95 signaling blocks butyrate induction of apoptosis in young adult mouse colonic cells. Am. J. Physiol. 1999, 277, C310–319. [Google Scholar]
- Terui, T.; Murakami, K.; Takimoto, R.; Takahashi, M.; Takada, K.; Murakami, T.; Minami, S.; Matsunaga, T.; Takayama, T.; Kato, J.; Niitsu, Y. Induction of PIG3 and NOXA through acetylation of p53 at 320 and 373 lysine residues as a mechanism for apoptotic cell death by histone deacetylase inhibitors. Cancer Res. 2003, 63, 8948–8954. [Google Scholar]
- Chopin, V.; Slomianny, C.; Hondermarck, H.; Le Bourhis, X. Synergistic induction of apoptosis in breast cancer cells by cotreatment with butyrate and TNF-alpha, TRAIL, or anti-Fas agonist antibody involves enhancement of death receptors’ signaling and requires P21(waf1). Exp. Cell Res. 2004, 298, 560–573. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, J.W.; Lee, J.Y.; Kwon, T.K. Sodium butyrate sensitizes TRAIL-mediated apoptosis by induction of transcription from the DR5 gene promoter through Sp1 sites in colon cancer cells. Carcinogenesis 2004, 25, 1813–1820. [Google Scholar] [CrossRef]
- Nakata, S.; Yoshida, T.; Horinaka, M.; Shiraishi, T.; Wakada, M.; Sakai, T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 2004, 23, 6261–6271. [Google Scholar] [CrossRef]
- Joseph, J.; Wajapeyee, N.; Somasundaram, K. Role of p53 status in chemosensitivity determination of cancer cells against histone deacetylase inhibitor sodium butyrate. Int. J. Cancer 2005, 115, 11–18. [Google Scholar] [CrossRef]
- Takimoto, R.; Kato, J.; Terui, T.; Takada, K.; Kuroiwa, G.; Wu, J.; Ohnuma, H.; Takahari, D.; Kobune, M.; Sato, Y.; Takayama, T.; Matsunaga, T.; Niitsu, Y. Augmentation of antitumor effects of p53 gene therapy by combination with HDAC inhibitor. Cancer Biol. Ther. 2005, 4, 421–428. [Google Scholar] [CrossRef]
- Krupitza, G.; Harant, H.; Dittrich, E.; Szekeres, T.; Huber, H.; Dittrich, C. Sodium butyrate inhibits c-myc splicing and interferes with signal transduction in ovarian carcinoma cells. Carcinogenesis 1995, 16, 1199–1205. [Google Scholar] [CrossRef]
- Velázquez, O.C.; Zhou, D.; Seto, R.W.; Jabbar, A.; Choi, J.; Lederer, H.M.; Rombeau, J.L. In vivo crypt surface hyperproliferation is decreased by butyrate and increased by deoxycholate in normal rat colon: associated in vivo effects on c-Fos and c-Jun expression. JPEN J. Parenter. Enteral. Nutr. 1996, 20, 243–250. [Google Scholar]
- Tang, S.J.; Huang, Y.M.; Wang, F.F. Analysis of c-fos expression in the butyrate-induced F-98 glioma cell differentiation. Biochem. J. 1995, 306 (Pt 1), 47–56. [Google Scholar]
- Bonnotte, B.; Favre, N.; Reveneau, S.; Micheau, O.; Droin, N.; Garrido, C.; Fontana, A.; Chauffert, B.; Solary, E.; Martin, F. Cancer cell sensitization to fas-mediated apoptosis by sodium butyrate. Cell Death Differ. 1998, 5, 480–487. [Google Scholar]
- Ogawa, K.; Yasumura, S.; Atarashi, Y.; Minemura, M.; Miyazaki, T.; Iwamoto, M.; Higuchi, K.; Watanabe, A. Sodium butyrate enhances Fas-mediated apoptosis of human hepatoma cells. J. Hepatol. 2004, 40, 278–284. [Google Scholar] [CrossRef]
- Emenaker, N.J.; Calaf, G.M.; Cox, D.; Basson, M.D.; Qureshi, N. Short-chain fatty acids inhibit invasive human colon cancer by modulating uPA, TIMP-1, TIMP-2, mutant p53, Bcl-2, Bax, p21 and PCNA protein expression in an in vitro cell culture model. J. Nutr. 2011, 131 (11 Suppl.), 3041S–3046S. [Google Scholar]
- Joseph, J.; Mudduluru, G.; Antony, S.; Vashistha, S.; Ajitkumar, P.; Somasundaram, K. Expression profiling of sodium butyrate (NaB)-treated cells: identification of regulation of genes related to cytokine signaling and cancer metastasis by NaB. Oncogene 2004, 23, 6304–6315. [Google Scholar] [CrossRef]
- Tong, X.; Yin, L.; Giardina, C. Butyrate suppresses Cox-2 activation in colon cancer cells through HDAC inhibition. Biochem. Biophys. Res. Commun. 2004, 317, 463–471. [Google Scholar] [CrossRef]
- Ammanamanchi, S.; Brattain, M.G. Restoration of transforming growth factor-beta signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J. Biol. Chem. 2004, 279, 32620–32625. [Google Scholar] [CrossRef]
- Perrin, P.; Cassagnau, E.; Burg, C.; Patry, Y.; Vavasseur, F.; Harb, J.; Le Pendu, J.; Douillard, J.Y.; Galmiche, J.P.; Bornet, F. An interleukin 2/sodium butyrate combination as immunotherapy for rat colon cancer peritoneal carcinomatosis. Gastroenterology 1994, 107, 1697–1708. [Google Scholar]
- Armstrong, F.; Mathers, J.C. Kill and cure: dietary augmentation of immune defences against colon cancer. Proc. Nutr. Soc. 2000, 59, 215–220. [Google Scholar] [CrossRef]
- Miller, S.J.; Zaloga, G.P.; Hoggatt, A.M.; Labarrere, C.; Faulk, W.P. Short-chain fatty acids modulate gene expression for vascular endothelial cell adhesion molecules. Nutrition 2005, 21, 740–748. [Google Scholar] [CrossRef]
- Yee, J.C.; de Leon Gatti, M.; Philp, R.J.; Yap, M.; Hu, W.S. Genomic and proteome exploration of CHO and hybridoma cells under sodium butyrate treatment. Biotechnol. Bioeng. 2007. [Google Scholar]
- Sawa, H.; Murakami, H.; Ohshima, Y.; Murakami, M.; Yamazaki, I.; Tamura, Y.; Mima, T.; Satone, A.; Ide, W.; Hashimoto, I.; Kamada, H. Histone deacetylase inhibitors such as sodium butyrate and trichostatin A inhibit vascular endothelial growth factor (VEGF) secretion from human glioblastoma cells. Brain Tumor Pathol. 2002, 19, 77–81. [Google Scholar] [CrossRef]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; Castronovo, V. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef]
- Sawa, H.; Murakami, H.; Kumagai, M.; Nakasato, M.; Yamauchi, S.; Matsuyama, N.; Tamura, Y.; Satone, A.; Ide, W.; Hashimoto, I.; Kamada, H. Histone deacetylase inhibitor, FK228, induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo. Acta Neuropathol. 2004, 107, 523–531. [Google Scholar] [CrossRef]
- Zgouras, D.; Becker, U.; Loitsch, S.; Stein, J. Modulation of angiogenesis-related protein synthesis by valproic acid. Biochem. Biophys. Res. Commun. 2004, 316, 693–697. [Google Scholar] [CrossRef]
- Heider, U.; Kaiser, M.; Sterz, J.; Zavrski, I.; Jakob, C.; Fleissner, C.; Eucker, J.; Possinger, K.; Sezer, O. Histone deacetylase inhibitors reduce VEGF production and induce growth suppression and apoptosis in human mantle cell lymphoma. Eur. J. Haematol. 2006, 76, 42–50. [Google Scholar] [CrossRef]
- Dong, X.F.; Song, Q.; Li, L.Z.; Zhao, C.L.; Wang, L.Q. Histone deacetylase inhibitor valproic acid inhibits proliferation and induces apoptosis in KM3 cells via downregulating VEGF receptor. Neuro Endocrinol. Lett. 2007, 28(6). [E-pub ahead of print]. [Google Scholar]
- Bar-Sela, G.; Jacobs, K.M.; Gius, D. Histone deacetylase inhibitor and demethylating agent chromatin compaction and the radiation response by cancer cells. Cancer J. 2007, 13, 65–69. [Google Scholar] [CrossRef]
- Arundel, C.M.; Glicksman, A.S.; Leith, J.T. Enhancement of radiation injury in human colon tumor cells by the maturational agent sodium butyrate (NaB). Radiat. Res. 1985, 104, 443–448. [Google Scholar] [CrossRef]
- Arundel, C.M.; Kenney, S.M.; Leith, J.T.; Glicksman, A.S. Contrasting effects of the differentiating agent sodium butyrate on recovery processes after x-irradiation in heterogeneous human colon tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 959–968. [Google Scholar] [CrossRef]
- Lopez, C.A.; Feng, F.Y.; Herman, J.M.; Nyati, M.K.; Lawrence, T.S.; Ljungman, M. Phenylbutyrate sensitizes human glioblastoma cells lacking wild-type p53 function to ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 214–220. [Google Scholar] [CrossRef]
- Entin-Meer, M.; Yang, X.; VandenBerg, S.R.; Lamborn, K.R.; Nudelman, A.; Rephaeli, A.; Haas-Kogan, D.A. In vivo efficacy of a novel histone deacetylase inhibitor in combination with radiation for the treatment of gliomas. Neuro Oncol. 2007, 9, 82–88. [Google Scholar] [CrossRef]
- Ueno, M.; Toyota, M.; Akino, K.; Suzuki, H.; Kusano, M.; Satoh, A.; Mita, H.; Sasaki, Y.; Nojima, M.; Yanagihara, K.; Hinoda, Y.; Tokino, T.; Imai, K. Aberrant methylation and histone deacetylation associated with silencing of SLC5A8 in gastric cancer. Tumour Biol. 2004, 25, 134–140. [Google Scholar] [CrossRef]
- Hong, C.; Maunakea, A.; Jun, P.; Bollen, A.W.; Hodgson, J.G.; Goldenberg, D.D.; Weiss, W.A.; Costello, J.F. Shared epigenetic mechanisms in human and mouse gliomas inactivate expression of the growth suppressor SLC5A8. Cancer Res. 2005, 65, 3617–3623. [Google Scholar] [CrossRef]
- Conley, B.A.; Egorin, M.J.; Tait, N.; Rosen, D.M.; Sausville, E.A.; Dover, G.; Fram, R.J.; Van Echo, D.A. Phase I study of the orally administered butyrate prodrug, tributyrin, in patients with solid tumors. Clin. Cancer Res. 1998, 4, 629–634. [Google Scholar]
- Patnaik, A.; Rowinsky, E.K.; Villalona, M.A.; Hammond, L.A.; Britten, C.D.; Siu, L.L.; Goetz, A.; Felton, S.A.; Burton, S.; Valone, F.H.; Eckhardt, S.G. A phase I study of pivaloyloxymethyl butyrate, a prodrug of the differentiating agent butyric acid, in patients with advanced solid malignancies. Clin. Cancer Res. 2002, 8, 2142–2148. [Google Scholar]
- Edelman, M.J.; Bauer, K.; Khanwani, S.; Tait, N.; Trepel, J.; Karp, J.; Nemieboka, N.; Chung, E.J.; Van Echo, D. Clinical and pharmacologic study of tributyrin: an oral butyrate prodrug. Cancer Chemother. Pharmacol. 2003, 51, 439–444. [Google Scholar]
- Reid, T.; Valone, F.; Lipera, W.; Irwin, D.; Paroly, W.; Natale, R.; Sreedharan, S.; Keer, H.; Lum, B.; Scappaticci, F.; Bhatnagar, A. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Lung Cancer 2004, 45, 381–386. [Google Scholar] [CrossRef]
- Cutts, S.M.; Rephaeli, A.; Nudelman, A.; Hmelnitsky, I.; Phillips, D.R. Molecular basis for the synergistic interaction of adriamycin with the formaldehyde-releasing prodrug pivaloyloxymethyl butyrate (AN-9). Cancer Res. 2001, 61, 8194–8202. [Google Scholar]
- Perrine, S.P.; Dover, G.H.; Daftari, P.; Walsh, C.T.; Jin, Y.; Mays, A.; Faller, D.V. Isobutyramide, an orally bioavailable butyrate analogue, stimulates fetal globin gene expression in vitro and in vivo. Br. J. Haematol. 1994, 88, 555–561. [Google Scholar] [CrossRef]
- Nudelman, A.; Gnizi, E.; Katz, Y.; Azulai, R.; Cohen-Ohana, M.; Zhuk, R.; Sampson, S.R.; Langzam, L.; Fibach, E.; Prus, E.; Pugach, V.; Rephaeli, A. Prodrugs of butyric acid. Novel derivatives possessing increased aqueous solubility and potential for treating cancer and blood diseases. Eur. J. Med. Chem. 2001, 36, 63–74. [Google Scholar] [CrossRef]
- Nudelman, A.; Rephaeli, A. Novel mutual prodrug of retinoic and butyric acids with enhanced anticancer activity. J. Med. Chem. 2000, 43, 2962–2966. [Google Scholar] [CrossRef]
- Rephaeli, A.; Rabizadeh, E.; Aviram, A.; Shaklai, M.; Ruse, M.; Nudelman, A. Derivatives of butyric acid as potential anti-neoplastic agents. Int. J. Cancer 1991, 49, 66–72. [Google Scholar] [CrossRef]
- Rephaeli, A.; Entin-Meer, M.; Angel, D.; Tarasenko, N.; Gruss-Fischer, T.; Bruachman, I.; Phillips, D.R.; Cutts, S.M.; Haas-Kogan, D.; Nudelman, A. The selectivty and anti-metastatic activity of oral bioavailable butyric acid prodrugs. Invest. New Drugs 2006, 24, 383–392. [Google Scholar] [CrossRef]
- Zimra, Y.; Nudelman, A.; Zhuk, R.; Rabizadeh, E.; Shaklai, M.; Aviram, A.; Rephaeli, A. Uptake of pivaloyloxymethyl butyrate into leukemic cells and its intracellular esterase-catalyzed hydrolysis. J. Cancer Res. Clin. Oncol. 2000, 126, 693–698. [Google Scholar]
- Nakase, H.; Okazaki, K.; Tabata, Y.; Uose, S.; Ohana, M.; Uchida, K.; Matsushima, Y.; Kawanami, C.; Oshima, C.; Ikada, Y.; Chiba, T. Development of an oral drug delivery system targeting immune-regulating cells in experimental inflammatory bowel disease: a new therapeutic strategy. J. Pharmacol. Exp. Ther. 2000, 292, 15–21. [Google Scholar]
- Lamprecht, A.; Ubrich, N.; Yamamoto, H.; Schäfer, U.; Takeuchi, H.; Maincent, P.; Kawashima, Y.; Lehr, C.M. Biodegradable nanoparticles for targeted drug delivery in treatment of inflammatory bowel disease. J. Pharmacol. Exp. Ther. 2001, 299, 775–781. [Google Scholar]
- Peart, M.J.; Smyth, G.K.; van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3697–3702. [Google Scholar] [CrossRef]
- Epping, M.T.; Wang, L.; Plumb, J.A.; Lieb, M.; Gronemeyer, H.; Brown, R.; Bernards, R. A functional genetic screen identifies retinoic acid signaling as a target of histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 17777–17782. [Google Scholar] [CrossRef]
- Lindemann, R.K.; Newbold, A.; Whitecross, K.F.; Cluse, L.A.; Frew, A.J.; Ellis, L.; Williams, S.; Wiegmans, A.P.; Dear, A.E.; Scott, C.L.; Pellegrini, M.; Wei, A.; Richon, V.M.; Marks, P.A.; Lowe, S.W.; Smyth, M.J.; Johnstone, R.W. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 8071–8076. [Google Scholar] [CrossRef]
- Fundarò, A.; Cavalli, R.; Bargoni, A.; Vighetto, D.; Zara, G.P.; Gasco, M.R. Non-stealth and stealth solid lipid nanoparticles (SLN) carrying doxorubicin: pharmacokinetics and tissue distribution after i.v. administration to rats. Pharmacol. Res. 2000, 42, 337–343. [Google Scholar] [CrossRef]
- Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C. Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 454–477. [Google Scholar] [CrossRef]
- Zara, G.P.; Cavalli, R.; Fundarò, A.; Bargoni, A.; Caputo, O.; Gasco, M.R. Pharmacokinetics of doxorubicin incorporated in solid lipid nanospheres (SLN). Pharmacol. Res. 1999, 40, 281–286. [Google Scholar] [CrossRef]
- Rich, J.N.; Bigner, D.D. Development of novel targeted therapies in the treatment of malignant glioma. Nat. Rev. Drug Discov. 2004, 3, 430–446. [Google Scholar] [CrossRef]
- Dehais, C.; Laigle-Donadey, F.; Marie, Y.; Kujas, M.; Lejeune, J.; Benouaich-Amiel, A.; Pedretti, M.; Polivka, M.; Xuan, K.H.; Thillet, J.; Delattre, J.Y.; Sanson, M. Prognostic stratification of patients with anaplastic gliomas according to genetic profile. Cancer 2006, 107, 1891–1897. [Google Scholar] [CrossRef]
- Sanson, M.; Laigle-Donadey, F.; Benouaich-Amiel, A. Molecular changes in brain tumors: prognostic and therapeutic impact. Curr. Opin. Oncol. 2006, 18, 623–630. [Google Scholar] [CrossRef]
- Carpentier, A.F.; Meng, Y. Recent advances in immunotherapy for human glioma. Curr. Opin. Oncol. 2006, 18, 631–636. [Google Scholar] [CrossRef]
- Gray, S.G.; Dangond, F. Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics 2006, 1, 67–75. [Google Scholar] [CrossRef]
- Boutillier, A.L.; Rouaux, C.; Pantaleeva, I.; Loeffler, J.Philippe. Chromatin acetylation status in the manifestation of neurodegenerative diseases: HDAC inhibitors as therapeutic tools. Subcell. Biochem. 2007, 41, 263–293. [Google Scholar]
- Morrison, B.E.; Majdzadeh, N.; D'Mello, S.R. Histone deacetylases: focus on the nervous system. Cell. Mol. Life Sci. 2007, 64, 2258–2269. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; Hersch, S.M. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar]
- Oliveira, J.M.; Chen, S.; Almeida, S.; Riley, R.; Gonçalves, J.; Oliveira, C.R.; Hayden, M.R.; Nicholls, D.G.; Ellerby, L.M.; Rego, A.C. Mitochondrial-dependent Ca2+ handling in Huntington's disease striatal cells: effect of histone deacetylase inhibitors. J. Neurosci. 2006, 26, 11174–11186. [Google Scholar] [CrossRef]
- Borovecki, F.; Lovrecic, L.; Zhou, J.; Jeong, H.; Then, F.; Rosas, H.D.; Hersch, S.M.; Hogarth, P.; Bouzou, B.; Jensen, R.V.; Krainc, D. Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc Natl. Acad. Sci. U.S.A. 2005, 102, 11023–11028. [Google Scholar] [CrossRef]
- Chang, J.G.; Hsieh-Li, H.M.; Jong, Y.J.; Wang, N.M.; Tsai, C.H.; Li, H. Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 9808–9813. [Google Scholar] [CrossRef]
- Minamiyama, M.; Katsuno, M.; Adachi, H.; Waza, M.; Sang, C.; Kobayashi, Y.; Tanaka, F.; Doyu, M.; Inukai, A.; Sobue, G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2004, 13, 1183–1192. [Google Scholar] [CrossRef]
- Mercuri, E.; Bertini, E.; Messina, S.; Solari, A.; D'Amico, A.; Angelozzi, C.; Battini, R.; Berardinelli, A.; Boffi, P.; Bruno, C.; Cini, C.; Colitto, F.; Kinali, M.; Minetti, C.; Mongini, T.; Morandi, L.; Neri, G.; Orcesi, S.; Pane, M.; Pelliccioni, M.; Pini, A.; Tiziano, F.D.; Villanova, M.; Vita, G.; Brahe, C. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 2007, 68, 51–55. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.