Michael Additions of Amines to Methyl Acrylates Promoted by Microwave Irradiation

Abstract
:Introduction
Results and Discussion
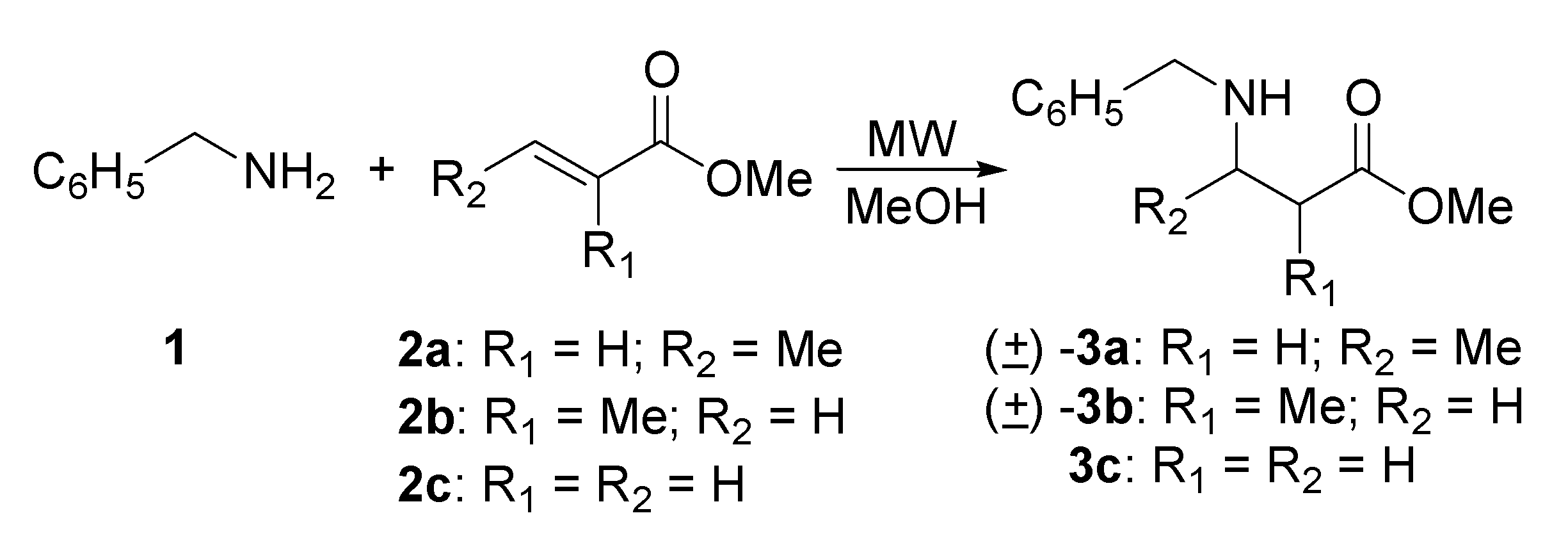
Preparation of N-benzyl-β-amino esters derived from benzylamine


{kind=link}
{kind=link}
{kind=link}
| Entry | Product | Power (Watts) | T (oC) | Pressure (psi) | Time (min) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | (±)-3a | 37 | 150 | 110 | 180 | 98 |
| 2 | (±)-3a | 55 | 140 | a | 180 | 83 |
| 3 | (±)-3b | 50 | 115-130 | 68 | 180 | 97 |
| 4 | (±)-3b | 55 | 140 | a | 390 | 80 |
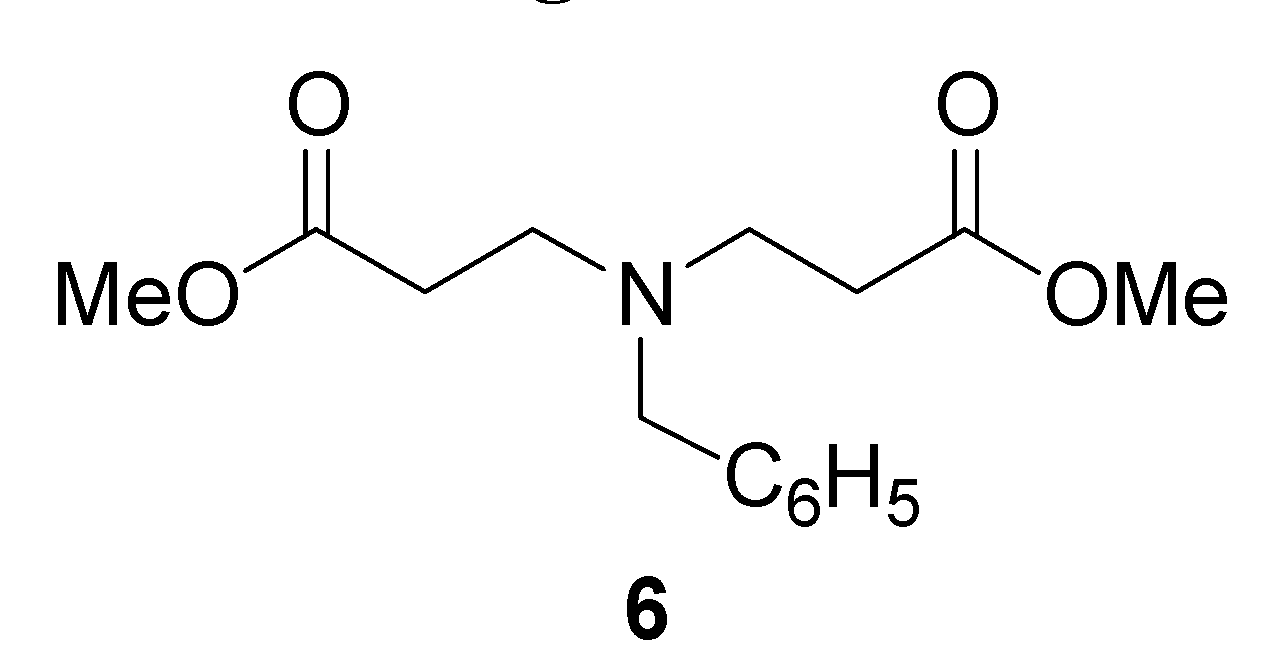
| 5 | 3c:6 | 50 | 115 | 59 | 180 | 50:50b |
| 6 | 3c:6 | 50 | 65 | 5 | 10 | 70:30b |
| 7 | 3c:6 | 40 | 65 | c | 10 | 70:30b |
| 8 | 3c:6 | 40 | 65 | c | 3 | 90:10b |

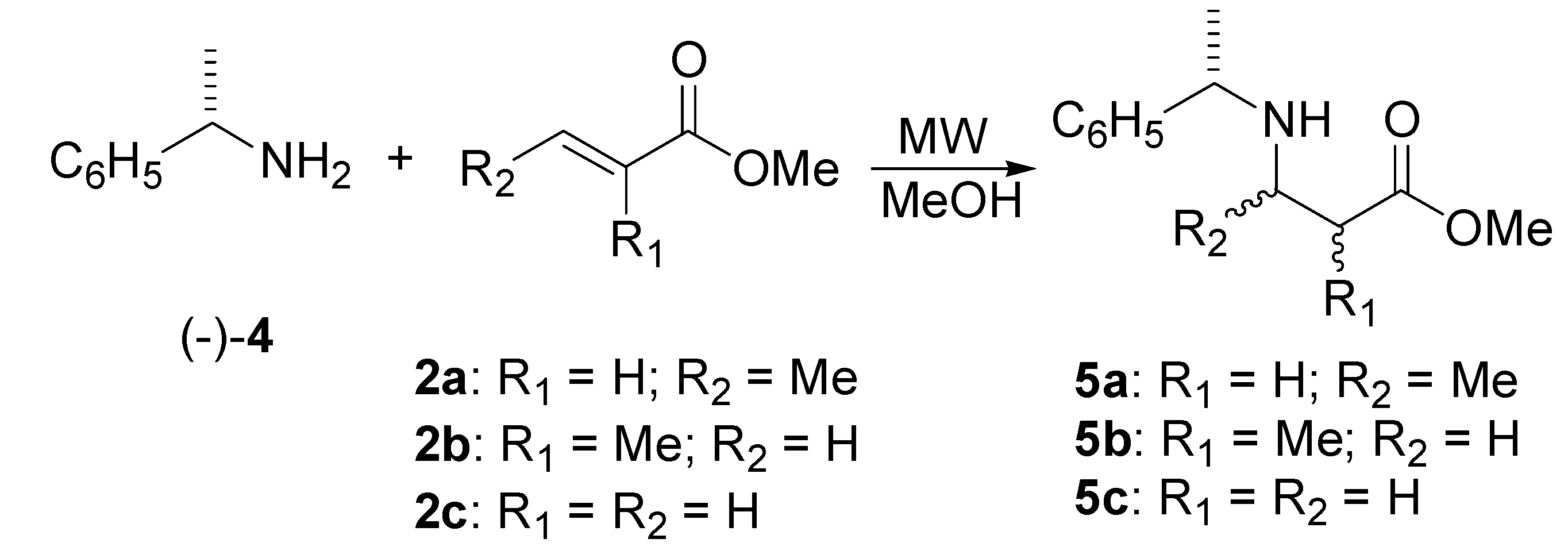
Preparation of β-amino esters derived from (S)-(-)-α-methylbenzylamine.

| Entry | Product | Power (Watts) | T (oC) | Pressure (psi) | Time (min) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 5a | 26 | 150 | 71 | 360 | 74 |
| 2 | 5b | 14 | 130-150 | 100 | 540 | 70 |
| 3 | 5b | 48 | 130-150 | a | 360 | 85 |
| 4 | 5c | 50 | 80 | 18 | 10 | 95 |
| 5 | 5c | 50 | 70 | b | 20 | quantitative |
Conclusions
Experimental
General
General procedure: Synthesis of N-benzyl-β-amino esters via microwave irradiation
(rac)-Methyl N-benzyl-3-aminobutanoate [(±)-3a]
(rac)-Methyl 3-(benzylamino)-2-methylpropanoate, [(±)-3b]
Methyl 3-(benzylamino)propanoate (3c) and bis [Methyl 3-(benzylamino)]propanoate (6)
Methyl N-α-Methylbenzylaminobutanoate (5a)
Methyl N-α-methylbenzyl-3-amino-2-methylpropionate (5b)
Methyl N-α-Methylbenzylaminopropanoate (5c)
Acknowledgements
References and Notes
- Hayes, B. L. Microwave Synthesis. Chemistry at the Speed of Light; CEM Publishing: Mattehews, NC, USA, 2002. [Google Scholar]
- For reviews see: Kappe, C. O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. [Google Scholar] Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis- a review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] Loupy, A.; Petit, A.; Hamelin, J.; Texier-Boullet, F.; Jacquault, P.; Mathé, D. New Solvent-Free Organic Synthesis Using Focused Microwaves. Synthesis 1998, 1213–1234. [Google Scholar] Caddick, S. Microwave Assisted Organic Reactions. Tetrahedron 1995, 51, 10403–10432. [Google Scholar]
- For other books see: Loupy, A. (Ed.) Microwaves in Organic Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2006. Tierney, J.P.; Lidström, P. (Eds.) Microwave Assisted Organic Synthesis; Blackwell Publishing: Oxford, UK, 2005. Kappe, C. O.; Stadler, A. Microwaves in Organic and Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Santra, S.; Andreana, R. A. One-Pot, Microwave-Influenced Synthesis of Diverse Small Molecules by Multicomponent Reaction Cascades. Org. Lett. 2007, 9, 5035–5038. [Google Scholar] [CrossRef]
- Sharma, A. K.; Gowdahalli, K.; Krzeminski, J.; Amin, S. Microwave-Assisted Suzuki Cross-Coupling Reaction, a Key Step in the Synthesis of Polycyclic Aromatic Hydrocarbons and Their Metabolites. J. Org. Chem. 2007, 72, 8987–8989. [Google Scholar] [CrossRef]
- Baxendale, I. R.; Lee, A.-I.; Ley, S. V. A Concise Synthesis of Carpanone Using Solid-Supported Reagents and Scavengers. J. Chem. Soc., Perkin Trans. 1 2002, 1850–1857. [Google Scholar] [CrossRef]
- Steinreiber, A.; Stadler, A.; Mayer, S. F.; Faber, K.; Kappe, C. O. High-Speed Microwave-Promoted Mitsunobu Inversions. Application Toward the Deracemization of Sulcatol. Tetrahedron Lett. 2001, 42, 6283–6286. [Google Scholar] [CrossRef]
- Amore, K. M.; Leadbeater, N. E.; Millar, T. A.; Schmink, J.R. Fast, easy, solvent-free, microwave-promoted Michael addition of anilines to α,β-unsaturated alkenes: synthesis of N-aryl functionalized β-amino esters and acids. Tetrahedron Lett 2006, 47, 8583–8586. [Google Scholar] Moghaddam, F. M.; Mohammadi, M.; Hosseinnia, A. Water promoted Michael Addition of Secondary Amines to α,β-unsaturated Carbonyl Compounds under Microwave Irradiation. Hosseini, M. Synth. Commun. 2000, 30, 643–650. [Google Scholar]
- Juaristi, E.; Soloshonok, V. A. (Eds.) Enantioselective Synthesis of β-Amino Acids, 2nd ed.; Wiley-VCH: New York, USA, 2005.
- Davies, S.G.; Garrido, N. M.; Kruchinin, D.; Ichihara, O.; Kotchie, L. J.; Price, P. D.; Price Mortimer, A. J.; Rusell, A. J.; Smith, A. D. Homochiral Lithium Amides for the Asymmetric Synthesis of β-amino Acids. Tetrahedron: Asymmetry 2006, 17, 1793–1811. [Google Scholar] Doi, H.; Sakai, T.; Iguchi, M.; Yamada, K.-I.; Tomioka, K. Chiral Ligand-Controlled Asymmetric Conjugate Addition of Lithium Amides to Enoates. J. Am. Chem. Soc. 2003, 125, 2886–2887. [Google Scholar] Davies, S. G.; Ichihara, O. Asymmetric Synthesis of R-β-Amino Butanoic Acid and S-β-tyrosine: Homochiral Lithium Amide Equivalents for Michael Additions to α,β-unsaturated Esters. Tetrahedron: Asymmetry 1991, 2, 183–186. [Google Scholar] Juaristi, E.; Quintana, D.; Escalante, J. Enantioselective Synthesis of β-Amino Acids. Aldrichim. Acta 1994, 27, 3–11. [Google Scholar]
- Gedey, S.; Liljeblad, A.; Fülöp, F.; Kanerva, L. T. Sequential resolution of ethyl 3-aminobutyrate with carboxylic acid esters by Candida antarctica lipase B. Tetrahedron: Asymmetry 1999, 10, 2573–2581. [Google Scholar] Gedey, S.; Liljeblad, A.; Lázár, L.; Fülöp, F.; Kanerva, L. T. Structural effects on chemo- and enantioselectivity of Candida antarctica lipase B - Resolution of β-amino esters. Can. J. Chem. 2002, 80, 565–570. [Google Scholar] Solymár, M.; Liljeblad, A.; Lázar, L.; Fülöp, F.; Kanerva, L. T. Lipase-catalysed kinetic resolution in organic solvents: an approach to enantiopure α-methyl-β-alanine esters. Tetrahedron: Asymmetry 2002, 13, 1923–1928. [Google Scholar]
- Flores-Sánchez, P.; Escalante, J.; Castillo, E. Enzymatic resolution of N-protected-β3-amino methyl esters, using lipase B from Candida Antarctica. Tetrahedron: Asymmetry 2005, 16, 629–634. [Google Scholar] [CrossRef]
- Seebach, D.; Estermann, H. Diastereoselektive alkylierung von 3-aminobutansäure in der 2-stellung. Helv. Chim. Acta 1988, 71, 1824–1839. [Google Scholar] Furukawa, M.; Okawara, T.; Terawaki, Y. Asymmetric Syntheses of b-amino acids by the addition of chiral amines to C=C double bond. Chem. Pharm. Bull. 1977, 25, 1319–1325. [Google Scholar]
- Shustov, G. V.; Rauk, A. 3-Methylazetidin-2-one and its precursors: Optical resolution and Absolute Configurations. Tetrahedron: Asymmetry 1996, 7, 699–708. [Google Scholar] [CrossRef]
- Sample Availability: Small samples (a few milligrams) of the compounds mentioned in this paper are available from the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Escalante, J.; Carrillo-Morales, M.; Linzaga, I. Michael Additions of Amines to Methyl Acrylates Promoted by Microwave Irradiation. Molecules 2008, 13, 340-347. https://doi.org/10.3390/molecules13020340
Escalante J, Carrillo-Morales M, Linzaga I. Michael Additions of Amines to Methyl Acrylates Promoted by Microwave Irradiation. Molecules. 2008; 13(2):340-347. https://doi.org/10.3390/molecules13020340
Chicago/Turabian StyleEscalante, Jaime, Manuel Carrillo-Morales, and Irma Linzaga. 2008. "Michael Additions of Amines to Methyl Acrylates Promoted by Microwave Irradiation" Molecules 13, no. 2: 340-347. https://doi.org/10.3390/molecules13020340
APA StyleEscalante, J., Carrillo-Morales, M., & Linzaga, I. (2008). Michael Additions of Amines to Methyl Acrylates Promoted by Microwave Irradiation. Molecules, 13(2), 340-347. https://doi.org/10.3390/molecules13020340