Prodrugs for Amines

Abstract
:Introduction
Prodrugs that rely (mostly) on enzymatic activation
N-Alkylation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
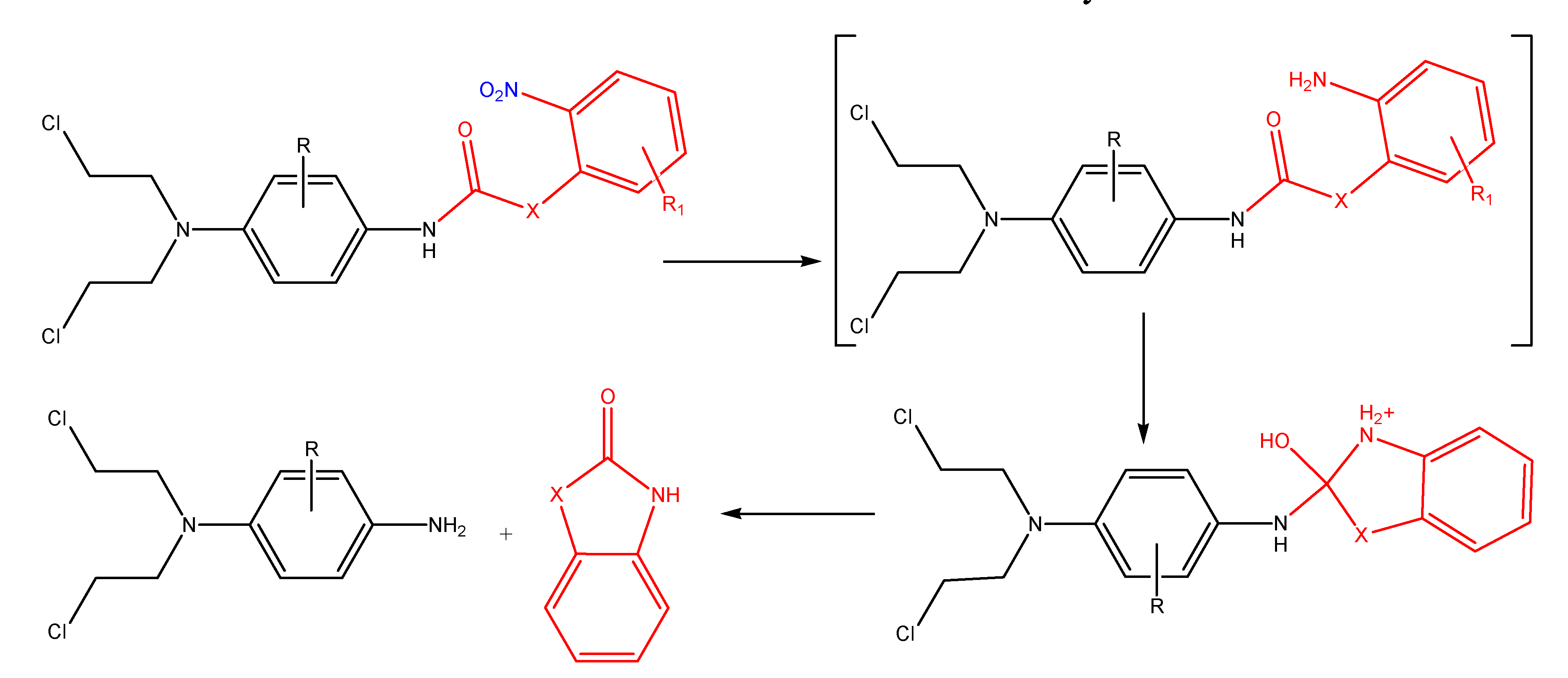
N-Acylation: amides and simple carbamates




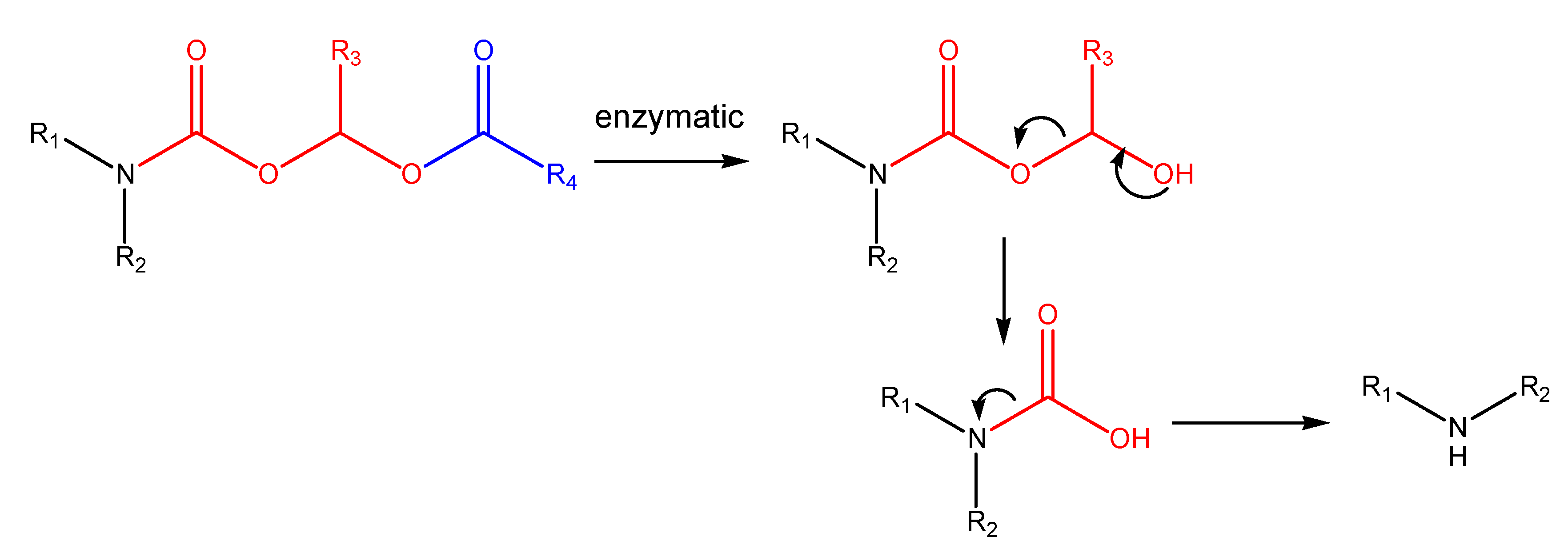
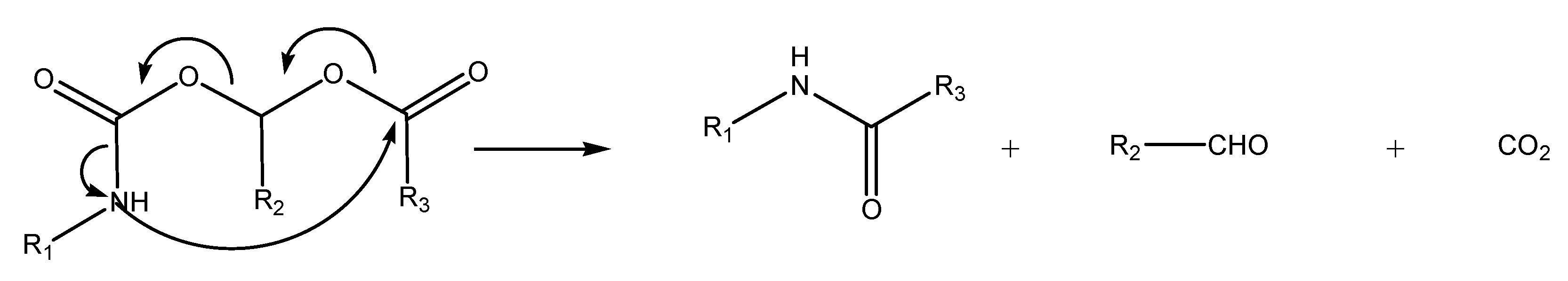
N-Acyloxyalkylation, N-hydroxyalkylation and N-(phosphoryloxy)alkylation



(Acyloxy)alkyl and (phosphoryloxy)alkyl carbamates




Azo compounds


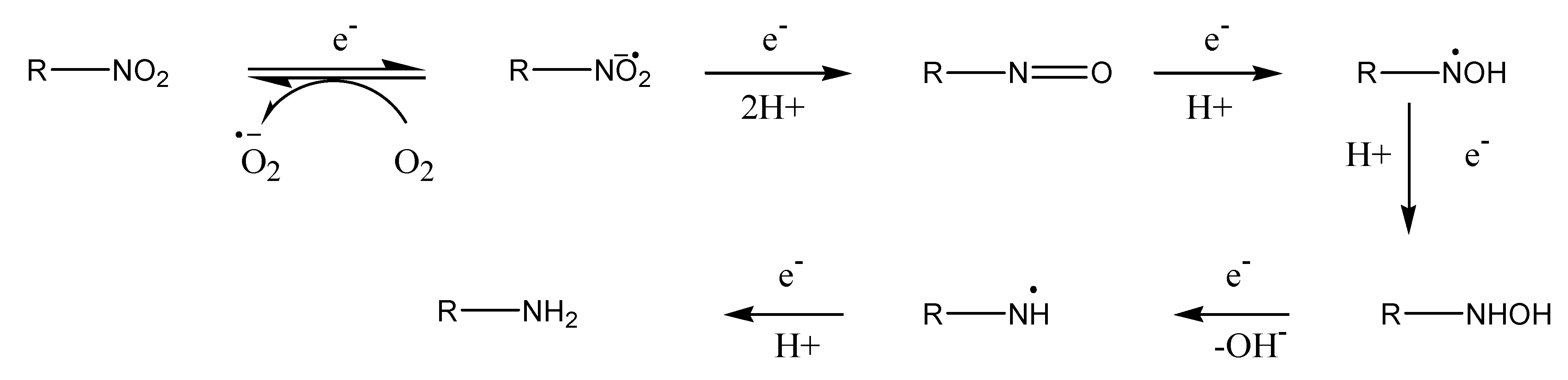
Redox systems






PEG and other macromolecular systems

Prodrugs that rely mostly on chemical activation
β-aminoketones

Schiff bases


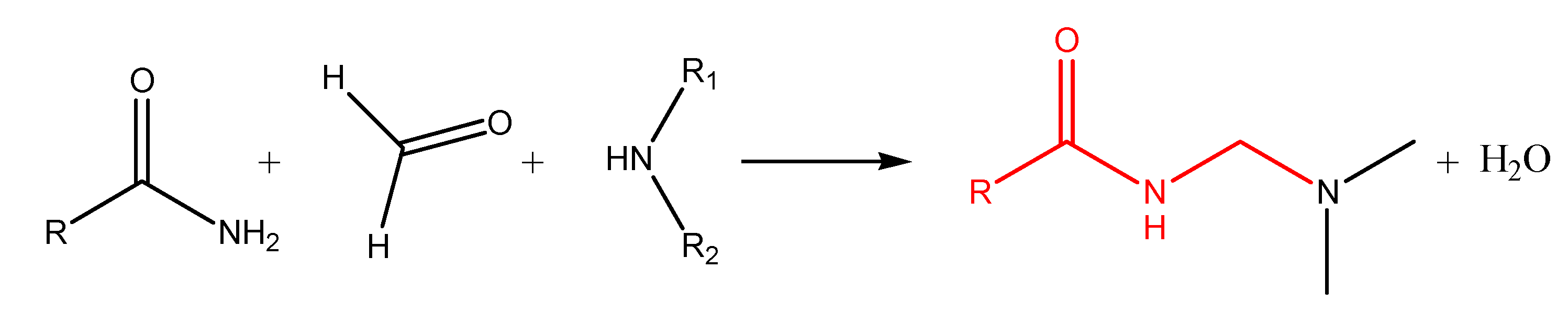
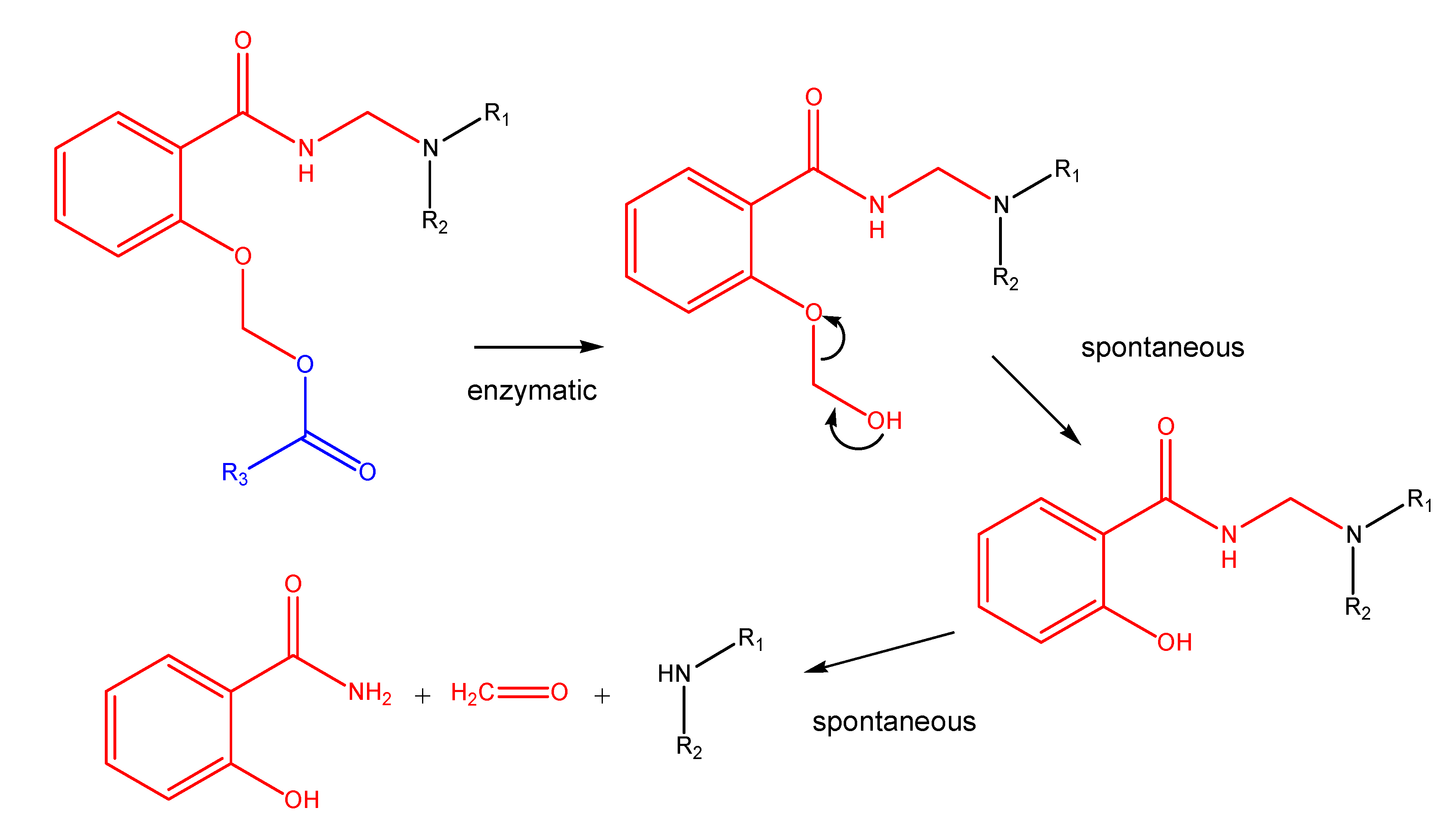
S and N-Mannich bases


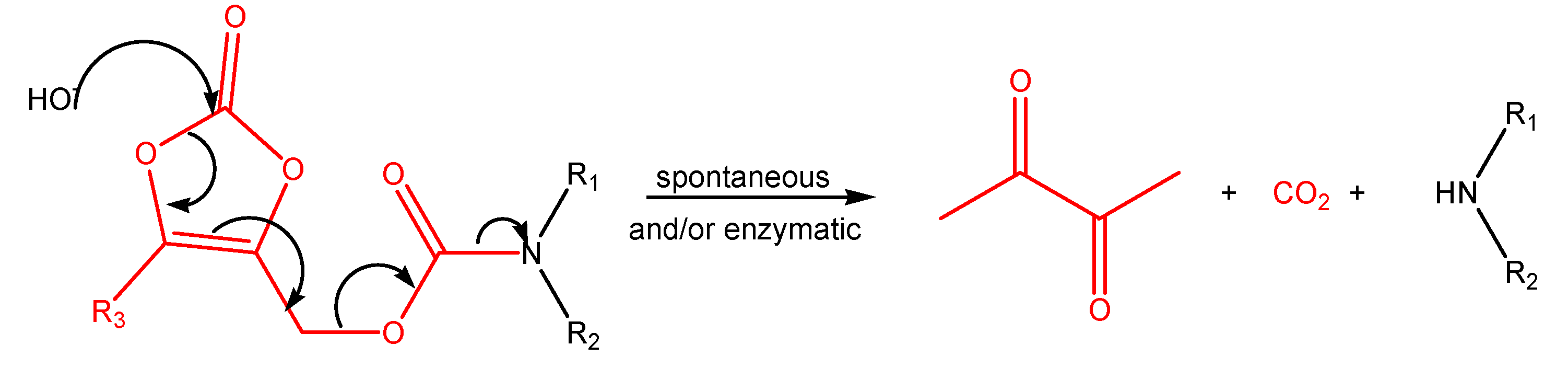
(Oxodioxolenyl)methyl carbamates and other selfimmolative systems


Enamines and enaminones


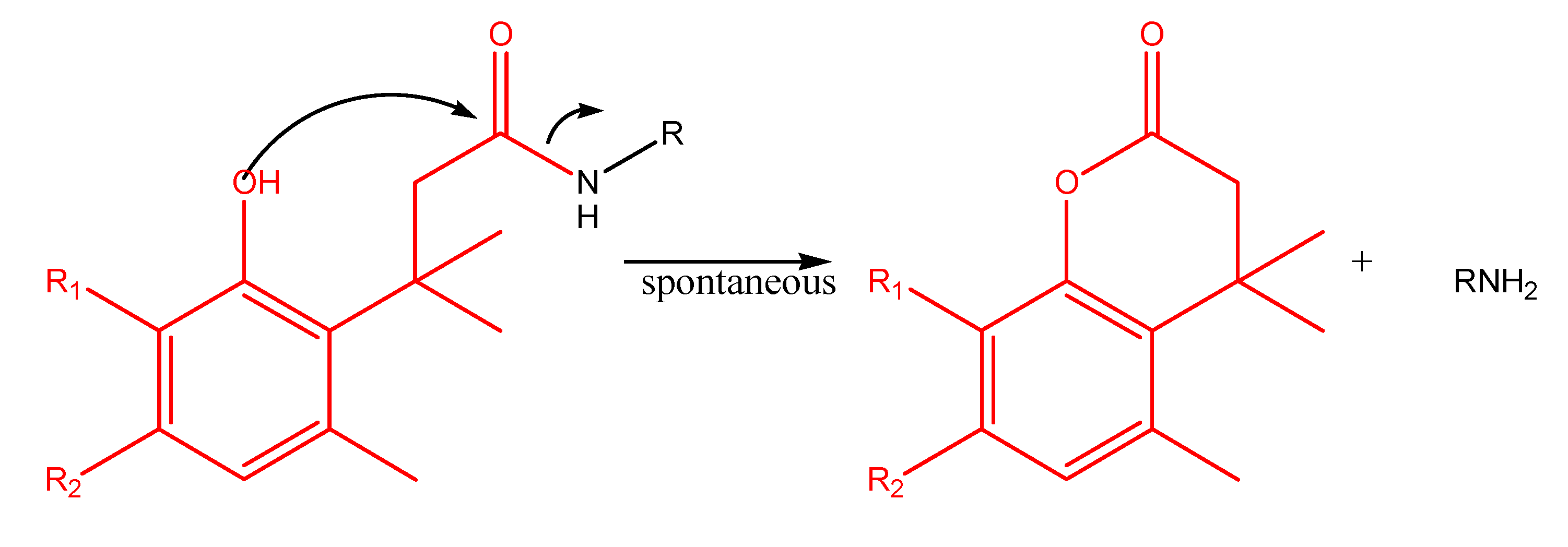
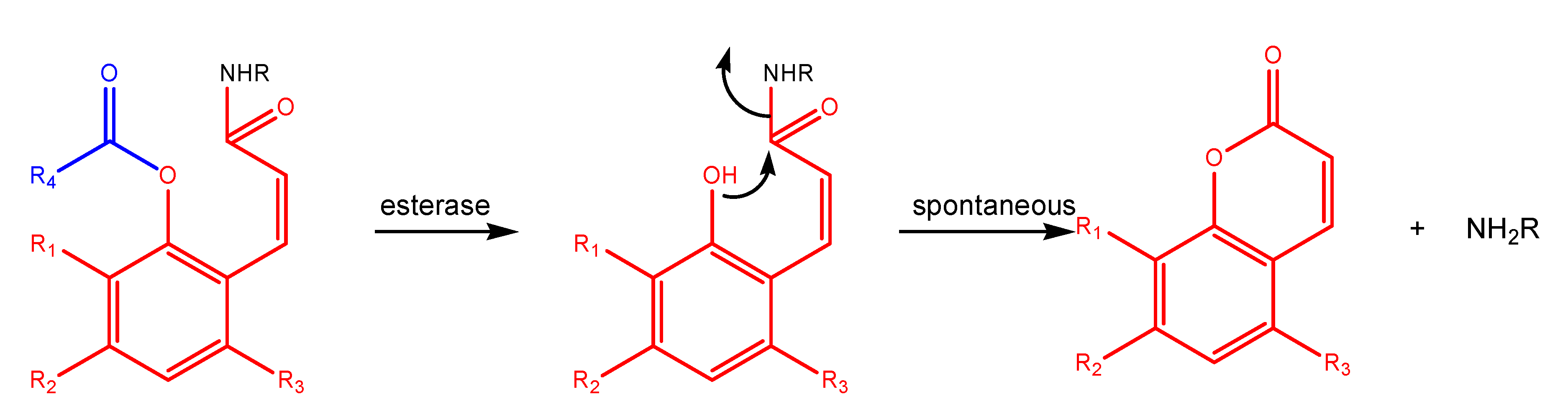
"Trimethyl lock" and coumarin systems



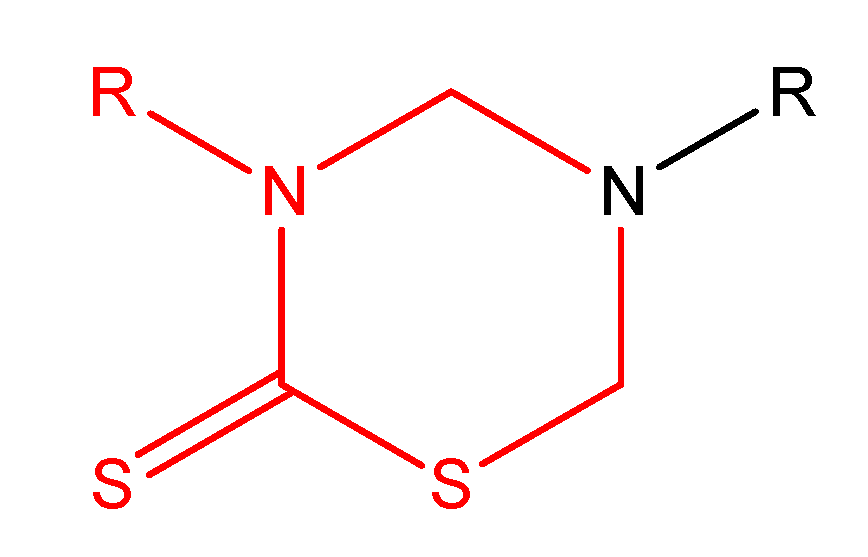
THTT

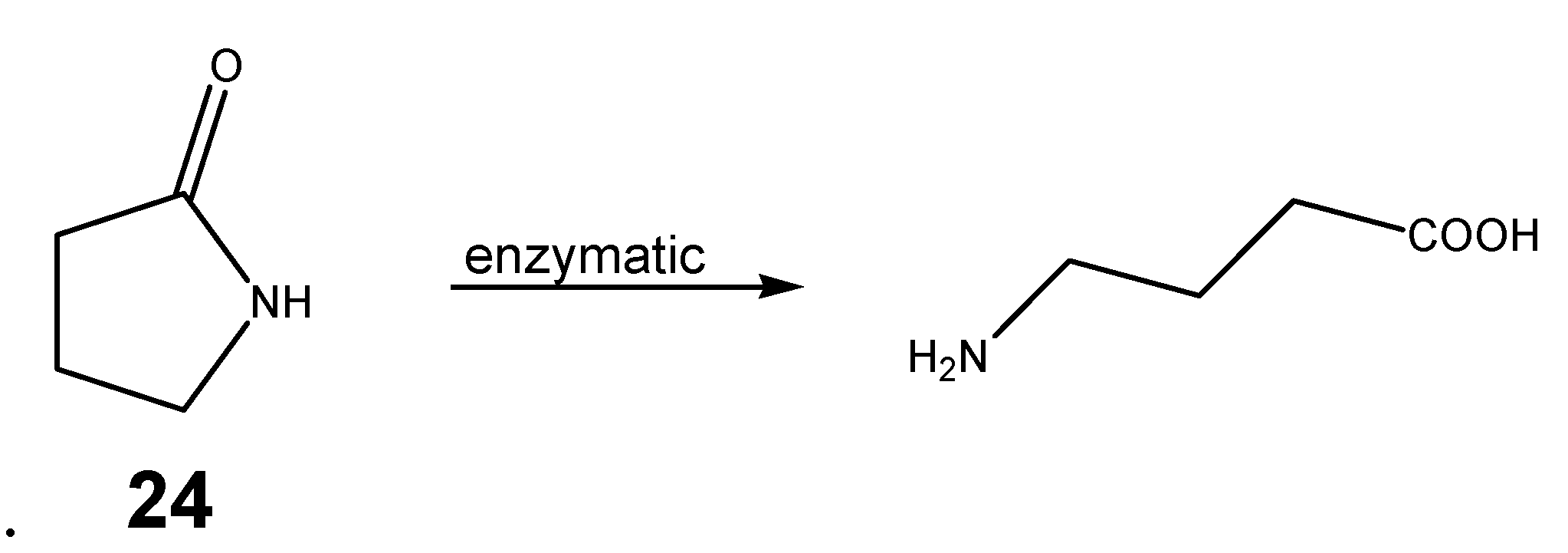
Cyclic derivatives of polyfunctional drugs containing the amino group



Conclusions
References
- Bundgaard, H.; Johansen, M. Prodrugs as drug delivery systems. XIX. Bioreversible derivatisation of aromatic amines by formation of N-Mannich bases with succinimide. Int. J. Pharm. 1981, 8, 183–192. [Google Scholar] [CrossRef]
- Bundgaard, H. Prodrugs as a means to improve the delivery of peptide drugs. 1. Adv. Drug Deliv. Rev. 1992, 8, 1–38. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, J.; Ballard, C.; Wang, B. Prodrug approaches to the improved delivery of peptide drugs. Curr. Pharm. Des. 1999, 5, 265–287. [Google Scholar]
- Gasparro, D.M.; Almeida, D.R.P.; Pisterzi, L.F.; Juhasz, J.R.; Viskolcz, B.; Penke, B.; Csizmadia, I.G. Reaction profiling of the MAO-B catalyzed oxidative deamination of amines in Alzheimer's disease. J. Mol. Struct.: THEOCHEM 2003, 666-667, 527–536. [Google Scholar] [CrossRef]
- Testa, B.; Mayer, J.M. Hydrolysis in Drug and Prodrug metabolism, Chemistry, biochemistry and enzymology.; Wiley-VCH: Weinheim, 2003; Chapter 4; p. 83. [Google Scholar]
- Rao, T.S.; Baker, G.B.; Coutts, R.T. N,N-dipropargyl-2-phenylethylamine, a potential prodrug of 2-phenylethylamine: neurochemical and neuropharmacological studies in rat. Brain Res. Bull. 1987, 19, 47–55. [Google Scholar] [CrossRef]
- Rao, T.S.; Baker, G.B.; Coutts, R.T. Pharmacokinetic and neurochemical studies on N-propargyl-2-phenylethylamine, a prodrug of 2-phenylethylamine. N.-S. Arch. Pharmacol. 1987, 336, 25–32. [Google Scholar]
- Baker, G.B.; Coutts, R.T.; Rao, T.S. Neuropharmacological and neurochemical properties of N-(2-cyanoethyl)-2-phenylethylamine, a prodrug of 2-phenylethylamine. Br. J. Pharmacol. 1987, 92, 243–255. [Google Scholar] [CrossRef]
- Rao, T.S.; Baker, G.B.; Coutts, R.T. N-(3-chloropropyl)phenylethylamine as a possible prodrug of beta-phenylethylamine: studies in the rat brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 1987, 11, 301–308. [Google Scholar] [CrossRef]
- Bundgaard, H. Design of prodrugs; Bundgaard, H., Ed.; Elsevier: Amsterdam, 1985; Chapter 1. [Google Scholar]
- Nishiyama, S.; Yoshikawa, M.; Yamaguchi, I. An orally effective peripheral dopamine prodrug - Docarpamine TA-870. Cardiovasc. Drug Rev. 1992, 10, 101–116. [Google Scholar] [CrossRef]
- Casagrande, C.; Merlo, L.; Ferrini, R.; Miragoli, G.; Semeraro, C. Cardiovascular and renal action of dopaminergic prodrugs. J. Cardiovasc. Pharmacol. 1989, 14, S40–S59. [Google Scholar] [CrossRef]
- Kearney, A.S. Prodrugs and targeted drug delivery. Adv. Drug Deliv. Rev. 1996, 19, 225–239. [Google Scholar] [CrossRef]
- Lee, M. Five years experience with γ-L-glutamyl-L-dopa- a relatively renally specific dopaminergic prodrug in man. J. Auton. Pharmacol. 1990, 10, S103–S108. [Google Scholar] [CrossRef]
- Matsubayashi, K.; Ueda, Y.; Ogino, H.; Sugita, T.; Sakakibara, Y.; Matsuyama, K.; Nomoto, T. Oral administration of the dopamine prodrug docarpamine shortens need for drip infusion of dopamine in patients with low cardiac output syndrome after cardiac surgery. Thorac. Cardiovasc. Surg. 1999, 47, 352–356. [Google Scholar]
- Tano, K.; Yoshizumi, M.; Kitagawa, T.; Hori, T.; Kitaichi, T.; Itoh, K.; Katoh, I. Effect of docarpamine, a novel orally active dopamine prodrug, on the formation of free and sulfoconjugated dopamine in patients who underwent cardiac surgery. Life Sci. 1997, 61, 1469–1478. [Google Scholar] [CrossRef]
- Nishiyama, S.; Yamaguchi, I.; Akimoto, Y.; Yoshikawa, M.; Nakajima, H. A novel orally active dopamine prodrug TA-870 II. Evidence that TA-870 is a dopamine prodrug. J. Cardiovasc. Pharmacol. 1989, 14, 175–183. [Google Scholar] [CrossRef]
- Carelli, V.; Liberatore, F.; Scipione, L.; Impicciatore, M.; Barocelli, E.; Cardellini, M.; Giorgioni, G. New systems for the specific and sustained release of dopamine to the brain. J. Control. Release 1996, 42, 209–216. [Google Scholar] [CrossRef]
- Pochopin, N.L.; Charman, W.N.; Stella, V.J. Pharmacokinetics of Dapsone and Amino Acid Prodrugs of Dapsone. Drug Metab. Dispos. 1994, 22, 770–775. [Google Scholar]
- Pochopin, N.L.; Charman, W.N.; Stella, V.J. Amino Acid Derivatives of Dapsone as Water-Soluble Prodrugs. Int. J. Pharm. 1995, 121, 157–167. [Google Scholar] [CrossRef]
- Bradshaw, T.D.; Bibby, M.C.; Double, J.A; Fichtner, I.; Cooper, P.A.; Alley, M.C.; Donohue, S.; Stinson, S.F.; Tomaszewjski, J.E.; Sausville, E.A.; Stevens, M.F. Preclinical evaluation of amino acid prodrugs of novel antitumor 2-(4-amino-3-methylphenyl)benzothiazoles. Mol. Cancer. Ther. 2002, 1, 239–246. [Google Scholar]
- Florvall, L.; Fagervall, I.; Larson, L.; Ross, S.B. Prodrugs of neuron-selective monoamine oxidase inhibitors: amino acid derivatives of 1-(4-aminophenyl)-2-aminopropanes. Eur. J. Med. Chem. 1999, 34, 137–151. [Google Scholar] [CrossRef]
- Tsuda, M.; Terada, T.; Irie, M.; Katsura, T.; Niida, A.; Tomita, K.; Fujii, N.; Inui, K.J. Transport characteristics of a novel peptide transporter 1 substrate, antihypotensive drug midodrine, and its amino acid derivatives. Pharmaco.l Exp. Ther. 2006, 318, 455–460. [Google Scholar] [CrossRef]
- Bundgaard, H.; Falch, E. Allopurinol prodrugs. I. Synthesis, stability and physicochemical properties of various N1-acyl allopurinol derivatives. Int. J. Pharm. 1985, 23, 223–237. [Google Scholar] [CrossRef]
- Stankoviov, M. Kinetics of hydrolysis of acetyl, valeroyl and nicotinoyl acyl derivatives of stobadine. Life Sci. 1999, 65, 2007–2010. [Google Scholar] [CrossRef]
- Gershonov, E.; Goldwaser, I.; Fridkin, M.; Shechter, Y. A novel approach for a water-soluble long acting insulin prodrug: design, preparation, and analysis of [(2-sulfo)-9-fluorenylmethoxycarbonyl]3-insulin. J. Med. Chem. 2000, 43, 2530–2537. [Google Scholar] [CrossRef]
- Shechter, Y.; Tsubery, H.; Fridkin, M. [2-sulfo)-9-fluorenylmethoxycarbonyl]3-exendin-4 a long acting glucose-lowering prodrug. Biochem. Biophys. Res. Commun. 2003, 305, 386–391. [Google Scholar] [CrossRef]
- Jordan, A.; Khan, T.; Malkin, H.; Osborn, H. Synthesis and analysis of urea and carbamate prodrugs as candidates for melanocyte-directed enzyme prodrug therapy (MDEPT). Bioorg. Med. Chem. 2002, 10, 2625–2633. [Google Scholar] [CrossRef]
- Hecker, S.J.; Calkins, T.; Price, M.E.; Huie, K.; Chen, S.; Glinka, T.W; Dudley, M.N. Prodrugs of Cephalosporin RWJ-333441 (MC-04,546) with improved aqueous solubility. Antimicrob. Agents Chemother. 2003, 47, 2043–2046. [Google Scholar] [CrossRef]
- Yumibe, N.; Huie, K.; Chen, K.J.; Snow, M.; Clement, R.P.; Cayen, M.N. Identification of human liver cytochrome P450 enzymes that metabolize the nonsedating antihistamine loratadine. Formation of descarboethoxyloratadine by CYP3A4 and CYP2D6. Biochem Pharmacol. 1996, 51, 165–172. [Google Scholar] [CrossRef]
- Monroe, E. Desloratadine for the treatment of chronic urticaria. Skin Therapy Lett 2012, 7, 1-2,5. [Google Scholar]
- Shimma, N.; Umeda, I.; Arasaki, M.; Murasaki, C.; Masubuchi, K.; Kohchi, Y.; Miwa, M.; Ura, M.; Sawada, N.; Tahara, H.; Kuruma, I.; Horii, I.; Ishitsuka, H. The design and synthesis of a new tumor-selective fluoropyrimidine carbamate, capecitabine. Bioorg Med Chem. 2000, 8, 1697–1706. [Google Scholar] [CrossRef]
- Ajani, J. Review of capecitabine as oral treatment of gastric, gastroesophageal, and esophageal cancers. Cancer 2006, 107, 221–231. [Google Scholar] [CrossRef]
- Barthel, B.L.; Torres, R.C.; Hyatt, J.L.; Edwards, C.C.; Hatfield, M.J.; Potter, P.M.; Koch, T.H. Identification of human intestinal carboxylesterase as the primary enzyme for activation of a doxazolidine carbamate prodrug. J. Med. Chem. 2008, 51, 298–304. [Google Scholar] [CrossRef]
- Järvinen, T.; Järvinen, K. Prodrugs for improved ocular drug delivery. Adv. Drug Deliv. Rev. 1996, 19, 203–224. [Google Scholar] [CrossRef]
- Kerr, D.; Roberts, W.; Tebbett, I.; Sloan, K. /-Alkylcarbonyloxymethyl prodrugs of theophylline: topical delivery of theophylline. Int. J. Pharm. 1998, 167, 37–48. [Google Scholar] [CrossRef]
- Krise, J.P.; Zygmunt, J.; Georg, G.I; Stella, V.J. Novel prodrug approach for tertiary amines: synthesis and preliminary evaluation of N-phosphonooxymethyl prodrugs. J. Med. Chem. 1999, 42, 3094–3100. [Google Scholar] [CrossRef]
- Krise, J.P.; Narisawa, S.; Stella, V.J. A novel prodrug approach for tertiary amines. 2. Physicochemical and in vitro enzymatic evaluation of N-phosphonooxymethyl prodrugs. J. Pharm. Sci. 1999, 88, 922–927. [Google Scholar] [CrossRef]
- Krise, J.P.; Charman, W.N.; Charman, S.A.; Stella, V.J. A novel prodrug approach for tertiary amines. 3. In vivo evaluation of two N-phosphonooxymethyl prodrugs in rats and dogs. J. Pharm. Sci. 1999, 88, 928–932. [Google Scholar]
- Stella, V.J. A Case for Prodrugs: Fosphenytoin. Adv. Drug Del. Rev. 1996, 19, 311–330. [Google Scholar] [CrossRef]
- Gogate, U.S.; Repta, A.J.; Alexander, J. N-(acyloxyalkoxycarbonyl) derivatives as potential prodrugs for amines. I. Kinetics and mechanism of degradation in aqueous solutions. Int. J. Pharm. 1987, 40, 235–248. [Google Scholar] [CrossRef]
- Gogate, U.S.; Repta, A.J. N-(acyloxyalkoxycarbonyl) derivatives as potential prodrugs for amines. II. Esterase-catalysed release of parent amines from model prodrugs. Int. J. Pharm. 1987, 40, 249–255. [Google Scholar] [CrossRef]
- Li, Z.; Bitha, P.; Lang, S.; Lin, Y. Synthesis of (alkoxycarbonyloxy)methyl, (acyloxy)methyl and (oxodioxolenyl)methyl carbamates as bioreversible prodrug moieties for amines. Bioorg. Med. Chem. Lett. 1997, 22, 2909–2912. [Google Scholar]
- Bodor, N. Design of prodrugs; Bundgaard, H., Ed.; Elsevier: Amsterdam, 1985; Chapter 11. [Google Scholar]
- Krogsgaard-Larsen, P.; Liljefors, T.; Madsen, U. A textbook of drug design and discovery, 3rd ed.; Taylor and Francis: New York, 2002; Chapter 14; pp. 410–458. [Google Scholar]
- Alexander, J.; Cargyl, R.; Michelson, S.; Schwam, H. (Acyloxy)alkyl carbamates as novel bioreversible prodrugs for amines: increased permeation through biological membranes. J. Med. Chem. 1988, 31, 318–322. [Google Scholar] [CrossRef]
- Alexander, J.; Fromtling, R.; Bland, J.; Pelak, B.; Gilfillan, E. (Acyloxy)alkyl carbamate prodrugs of norfloxacin. J. Med. Chem. 1991, 34, 78–81. [Google Scholar] [CrossRef]
- Cundy, K.C.; Branch, R.; Chernov-Rogan, T.; Dias, T.; Estrada, T.; Hold, K.; Koller, K.; Liu, X.; Mann, A.; Panuwat, M.; Raillard, S.P.; Upadhyay, S.; Wu, Q.Q.; Xiang, J.N.; Yan, H.; Zerangue, N.; Zhou, C.X.; Barrett, R.W.; Gallop, M.A. XP13512 [(+/-)-1-([(α-isobutanoyl-oxyethoxy)carbonyl]aminomethyl)-1-cyclohexane acetic acid], a Novel Gabapentin Prodrug: I. Design, Synthesis, Enzymatic Conversion to Gabapentin, and Transport by Intestinal Solute Transporters. J. Pharmacol. Exp. Ther. 2004, 311, 315–323. [Google Scholar] [CrossRef]
- Cundy, K.C.; Annamalai, T.; Bu, L.; De Vera, J.; Estrela, J.; Luo, W.; Shirsat, P.; Torneros, A.; Yao, F.; Zou, J.; Barrett, R.W.; Gallop, M.A. XP13512 [(+/-)-1-([(α-isobutanoyl-oxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a Novel Gabapentin Prodrug: II. Improved Oral Bioavailability, Dose Proportionality, and Colonic Absorption Compared with Gabapentin in Rats and Monkeys. J. Pharmacol. Exp. Ther. 2004, 311, 324–333. [Google Scholar] [CrossRef]
- Alexander, J.; Bindra, D.; Glass, J.; Holahan, M.; Reyner, M.; Rork, G.; Sitko, G.; Stranieri, M.; Stupienski, R.; Veerpanane, H.; Cook, J. Investigation of (oxodioxolenyl)methyl carbamates as nonchiral bioreversible prodrug moieties for chiral amines. J. Med. Chem. 1996, 39, 480–486. [Google Scholar] [CrossRef]
- Safadi, M.; Oliyai, R.; Stella, V.J. Phosphoryloxymethyl carbamates and carbonates - novel water soluble prodrugs for amines and hindered alcohols. Pharm. Res. 1993, 10, 1350–1355. [Google Scholar] [CrossRef]
- Kirshner, J. The origin of 20th century discoveries transforming clinical gastroenterology. Am. J. Gastroenterol. 1998, 93, 862–871. [Google Scholar]
- Sandborn, W. Rational selection of oral 5-aminosalicylate formulations and prodrugs for the treatment of ulcerative colitis. Am. J. Gastroenterol. 2002, 97, 2939–2941. [Google Scholar] [CrossRef]
- Jung, Y.; Lee, J.; Kim, Y. Colon-specific prodrug of 5-aminosalicylic acid: synthesis and in vitro/in vivo properties of acidic amino acid derivatives of 5-aminosalicylic acid. J. Pharm. Sci. 2001, 90, 1767–1775. [Google Scholar] [CrossRef]
- Rauth, A.; Melo, T.; Misra, V. Bioreductive therapies: an overview of drugs and their mechanism of action. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 755–762. [Google Scholar] [CrossRef]
- Everett, S.; Swann, E.; Naylor, M.; Stratford, M.; Patel, K.; Tian, N.; Newman, R.; Vojnovic, B.; Moody, C.; Wardman, P. Modifying rates of reductive elimination of leaving groups from indolequinone prodrugs: a key factor in controlling hypoxia-selective drug release. Biochem. Pharmacol. 2002, 63, 1629–1639. [Google Scholar] [CrossRef]
- Atwell, G.; Sykes, B.; O'Connor, C.; Denny, W. Relationships between structure and kinetics of cyclization-activated aromatic mustards. J. Med. Chem. 1994, 37, 371–380. [Google Scholar] [CrossRef]
- Guise, C.P.; Wang, A.T.; Theil, A.; Bridewell, D.J.; Wilson, W.R.; Patterson, A.V. Identification of human reductases that activate the dinitrobenzamide mustard prodrug PR-104A: a role for NADPH:cytochrome P450 oxidoreductase under hypoxia. Biochem. Pharmacol. 2007, 74, 810–820. [Google Scholar] [CrossRef]
- Sykes, B.; Atwell, G.; Hogg, A.; Wilson, W.; O'Connor, C.; Denny, W. N-substituted 2-(2,6-dinitrophenylamino)propanamides: novel prodrugs that release a primary amine via nitroreduction and intramolecular cyclization. J. Med. Chem. 1999, 42, 346–355. [Google Scholar] [CrossRef]
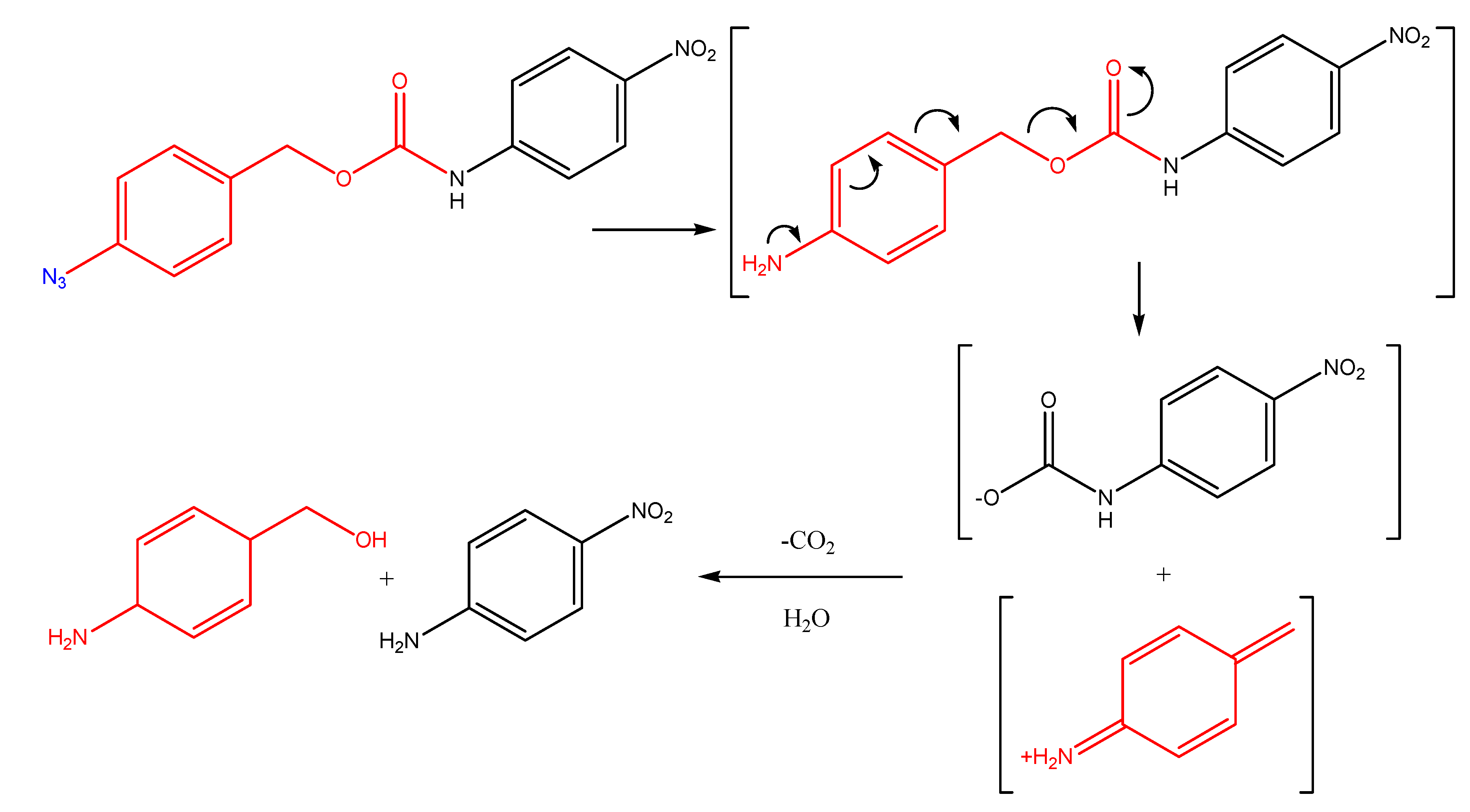
- Griffin, R.; Evers, E.; Davison, R.; Gibson, A.; Layton, D.; Irwin, W. The 4-azidoberazyloxycarbonyl function: application as a novel protecting group and potential prodrug modification for amines. J. Chem. Soc. Perkin Trans. 1 1996, 11, 1205–1211. [Google Scholar]
- Wilson, W.; Denny, W.; Pullen, S.; Thompson, K.; Li, A.; Patterson, L.; Lee, H. Tertiary amines N-oxides as bioreductive drugs: DACA N-oxide, nitracarine N-oxide and AQ4N. Br. J. Cancer 1996, 27, S43–S47. [Google Scholar]
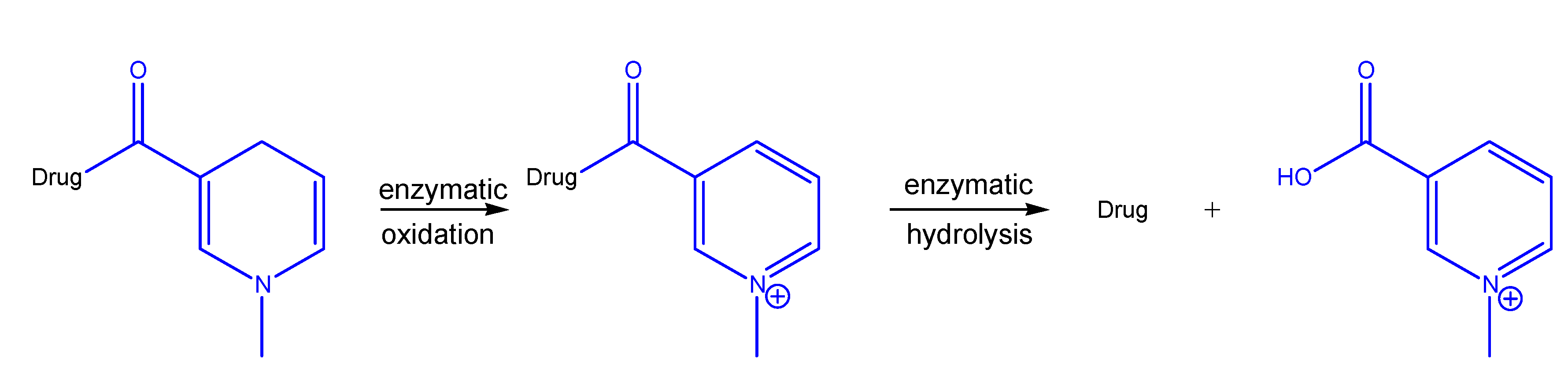
- Bodor, N.; Sheck, E.; Higuchi, T. Delivery of a quaternary pyridinium salt across the blood-brain barrier by its dihydropyridine derivative. Science 1975, 190, 155–156. [Google Scholar] [CrossRef]
- Bodor, N.; Farag, H.; Brewster, M. Site-specific, sustained release of drugs to the brain. Science 1981, 217, 1370–1372. [Google Scholar]
- Pop, E.; Wu, M.; Shek, E.; Bodor, N. Chemical delivery systems for drugs containing an amino group: synthesis and properties of some pyridine derivatives of desipramine. Drug. Des. Deliv. 1989, 5, 93–115. [Google Scholar]
- Bodor, N.; Simpkins, J. Redox delivery system for brain-specific, sustained release of dopamine. Science 1983, 221, 65–67. [Google Scholar]
- Simpkins, J.; Bodor, N.; Enz, A. Direct evidence for brain-specific release of dopamine from a redox delivery system. J. Pharm. Sci. 1985, 10, 1033–1036. [Google Scholar] [CrossRef]
- Brewster, M.; Bodor, N. Improved brain delivery of antiviral agents through the use of redox targeting. Adv. Drug Deliv. Rev. 1994, 14, 177–197. [Google Scholar] [CrossRef]
- Pop, E.; Sot, F.; Anderson, W.; Panetta, J.; Estes, K.; Bodor, N.; Brewster, M. Redox targeting of LY231617, an antioxidant with potential use in the treatment of brain damage. Int. J. Pharm. 1996, 140, 33–44. [Google Scholar] [CrossRef]
- Prokai, L.; Prokai-Tatri, L.; Bodor, N. Targeting drug to the brain by redox chemical delivery systems. Med. Res. Rev. 2000, 20, 367–416. [Google Scholar] [CrossRef]
- Ishikura, T.; Senou, T.; Ishiara, H.; Kato, T.; Ito, T. Drug delivery to the brain. DOPA prodrugs based on a ring-closure reaction to quaternary thiazolium compounds. Int. J. Pharm. 1995, 116, 51–63. [Google Scholar] [CrossRef]
- Sheha, M.; Al-Tayeb, A.; El-Sherief, H.; Farag, H. New carrier for specific delivery of drugs to the brain. Bioorg. Med. Chem. 2003, 11, 1865–1872. [Google Scholar] [CrossRef]
- Greenwald, R.; Pendri, A.; Conover, C.; Zhao, H.; Choe, Y.; Martinez, A.; Shum, K.; Guan, S. Drug delivery systems employing 1,4- or 1,6-elimination: poly(ethylene glycol) prodrugs of amine-containing compounds. J. Med. Chem. 1999, 42, 3657–3667. [Google Scholar] [CrossRef]
- Greenwald, R. PEG drugs: an overview. J. Control. Release 2001, 74, 159–171. [Google Scholar] [CrossRef]
- Greenwald, R.; Choe, Y.; Conover, C.; Shum, K.; Wu, D.; Royzen, M. Drug delivery systems based on trimethyl lock lactonization: poly(ethylene glycol) prodrugs of amine-containing compounds. J. Med. Chem. 2000, 43, 475–487. [Google Scholar] [CrossRef]
- Greenwald, R.; Zhao, H.; Xia, J. Tripartate poly(ethylene glycol) prodrugs of the open lactone form of camptothecin. Bioorg. Med. Chem. 2003, 11, 2635–2639. [Google Scholar] [CrossRef]
- Greenwald, R.; Choe, Y.; Mcguire, J.; Conover, C. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Zhao, H. Prodrugs: Challenges and Rewards Part I; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer: New York, 2007; Chapter 2.3.1; pp. 283–337. [Google Scholar]
- Franssen, E.; Koiter, J.; Kuipers, C.; Bruins, A.; Moolenaar, F.; Zeeuw, D.; Kruizinga, W.; Kellog, R.; Meijer, D. Low molecular weight proteins for renal drug targeting. Preparation of Drug-protein conjugates and drug-spacer derivatives and their catabolism in renal cortex homogenates and lysosomal lysates. J. Med. Chem. 1992, 35, 1246–1459. [Google Scholar] [CrossRef]
- Takakura, Y; Matsumoto, S.; Hashida, M.; Sezaki, H. Physicochemical Propertiesband Antitumor Activities of Polymeric Prodrugs of Mitomycin C with Different Regeneration Rates. J. Control. Release 1989, 10, 97–105. [Google Scholar] [CrossRef]
- Gilmer, J.F.; Simplício, A.L.; Clancy, J.C. A new amino-masking group capable of pH-triggered amino-drug release. Eur. J. Pharm. Sci. 2005, 24, 315–323. [Google Scholar] [CrossRef]
- Gilmer, J.F.; Simplício, A.L.; Clancy, J.C. β-Aminoketones as prodrugs with pH-controlled activation. Int. J. Pharm. 2007, 336, 208–214. [Google Scholar] [CrossRef]
- Krause, M.; Rouleau, A.; Stark, H.; Luger, P.; Lipp, R.; Garbarg, M.; Schwartz, J-C.; Schunack, W. Synthesis, X-ray christalography and pharmacokinetics of novel azomethine prodrugs of R-α-methylhistamine: highly potent and selective histamine H3-receptor agonists. J. Med. Chem. 1995, 38, 4070–4079. [Google Scholar] [CrossRef]
- Krause, M.; Stark, H.; Schunack, W. Azomethine prodrugs of R-α-methylhistamine, a highly potent and selective histamine H3-receptor agonist. Curr. Med. Chem. 2001, 8, 1329–1340. [Google Scholar] [CrossRef]
- Stark, H.; Krause, M.; Rouleau, A.; Garbarg, M.; Schwartz, J-C.; Schunack, W. Enzyme-catalyzed prodrug approaches for the histamine H3-receptor agonist R-α-methylhistamine. Bioorg. Med. Chem. 2001, 9, 191–198. [Google Scholar] [CrossRef]
- Bundgaard, H.; Johansen, M. Prodrugs as drug delivery systems IV: N-mannich bases as potential novel prodrugs for amides, ureides, amines, and other NH-acidic compounds. J. Pharm. Sci. 1980, 69, 44–46. [Google Scholar] [CrossRef]
- Higuchi, T. Bioreversible Carriers in Drug Design; Roche, E.B., Ed.; Pergamon Press: New York, 1987. [Google Scholar]
- Johansen, M.; Bundgaard, H. Pro-drugs as drug delivery systems. XIII. Kinetics of decomposition of N-Mannich bases of salicylamide and assessment of their suitability as possible pro-drugs for amines. Int. J. Pharm. 1980, 7, 119–127. [Google Scholar] [CrossRef]
- Bundgaard, H.; Lixbüll, U.; Falch, E. Prodrugs as drug delivery systems. 43. O-acyloxymethyl salicylamide N-Mannich bases as double prodrug forms for amines. Int. J. Pharm. 1986, 29, 19–28. [Google Scholar] [CrossRef]
- Bundgaard, H.; Johansen, M. Hydrolysis of N-Mannich bases and its consequences for the biological testing of such agents. Int. J. Pharm. 1981, 9, 7–16. [Google Scholar] [CrossRef]
- Johansen, M.; Bundgaard, H. Pro-drugs as drug delivery systems. XXIV. N-Mannich bases as bioreversible lipophilic transport forms for ephedrine, phenethylamine and other amines. Arch. Pharm. Chemi. Sci. Ed. 1982, 10, 111–115. [Google Scholar]
- Testa, B.; Mayer, J.M. Hydrolysis in Drug and Prodrug metabolism, Chemistry, biochemistry and enzymology; Wiley-VCH: Weinheim, 2003; Chapter 11; pp. 690–695. [Google Scholar]
- Sun, C.; Rodriguez, M.; Zeckner, D.; Sachs, B.; Current, W.; Boyer, R.; Paschal, J.; McMillian, C.; Chen, S. Synthesis and evaluation of oxodioxolenylmethyl carbamate prodrugs of pseudomycins. J. Med. Chem. 2001, 44, 2671–2674. [Google Scholar] [CrossRef]
- Erez, R.; Ebner, S.; Attali, B.; Shabat, D. Chemotherapeutic bone-targeted bisphosphonate prodrugs with hydrolytic mode of activation. Bioorg. Med. Chem. Lett. 2008, 18, 816–820. [Google Scholar] [CrossRef]
- Prats, G.; Rossi, V.; Salvatori, E.; Mirelis, B. Prulifloxacin: a new antibacterial fluoroquinolone. Expert Rev. Anti. Infect. Ther. 2006, 4, 27–41. [Google Scholar] [CrossRef]
- Tougou, K.; Nakamura, A.; Watanabe, S.; Okuyama, Y.; Morino, A. Paraoxonase has a major role in the hydrolysis of prulifloxacin (NM441), a prodrug of a new antibacterial agent. Drug. Metab. Dispos. 1998, 26, 355–359. [Google Scholar]
- Caldwell, H.; Adams, H.; Jones, R.; Mann, W.; Dittert, L.; Chang, C.; Swintosky, J. Enamine prodrugs. J. Pharm. Sci. 1971, 60, 1810–1812. [Google Scholar] [CrossRef]
- Murakami, T.; Tamauchi, H.; Yamazaki, M.; Kubo, K.; Kamada, A.; Yata, K. Biopharmaceutical study on the oral and rectal administration of enamine prodrugs of amino acid-like β-lactam antibiotics in rabbits. Chem. Pharm. Bull. 1981, 29, 1986–1997. [Google Scholar] [CrossRef]
- Dixon, K.; Greenhill, J. A study of the rates of hydrolysis of certain enaminones. J. Chem. Soc. Perkin. Trans. 1I 1974, 2, 164–168. [Google Scholar] [CrossRef]
- Naringrekar, V.; Stella, V.J. Mechanism of hydrolysis and structure-stability relationship of enaminones as potential prodrugs of model primary amines. J. Pharm. Sci. 1990, 79, 138–146. [Google Scholar] [CrossRef]
- Nielsen, N.; Bundgaard, H. Prodrugs as drug delivery systems. 42. 2-Hydroxymethylbenzamides and 2-acyloxymethylbenzamides as potential prodrug forms for amines. Int. J. Pharm. 1986, 29, 9–18. [Google Scholar] [CrossRef]
- Wang, B.; Nicolaou, M.; Liu, S.; Borchardt, R.T. Structural Analysis of a facile lactonization system facilitated by a "trimethyl lock". Bioorg. Chem. 1996, 24, 39–49. [Google Scholar] [CrossRef]
- Ameberry, K.; Gerstenberger, A.; Borchardt, R.T. Amine prodrugs which utilize hydroxy amide lactonization. II. A potential esterase-sensitive amide prodrug. Pharm. Res. 1991, 8, 455–461. [Google Scholar] [CrossRef]
- Amsberry, K.; Borchardt, R.T. Amine prodrugs which utilize hydroxy amide lactonization. I. A potential redox-sensitive amide prodrug. Pharm. Res. 1991, 8, 323–330. [Google Scholar] [CrossRef]
- Nicolaou, M.; Yuan, C.; Borchardt, R.T. Phosphate prodrugs for amines utilizing a fast intramolecular hydroxy amide lactonization. J. Org. Chem. 1996, 61, 8636–8641. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, H.; Wang, W. Chemical feasibility studies of a potential coumarin-based prodrug system. Bioorg. Med. Chem. Lett. 1996, 6, 945–950. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, H.; Zheng, A.; Wang, W. Coumarin-based prodrugs. Part 3: structural effects on the release kinetics of esterase-sensitive prodrugs for amines. Bioorg. Med. Chem. 1998, 6, 417–426. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, B. Substituted coumarins as esterase-sensitive prodrug moieties with improved release rates. Bioorg. Med. Chem. Lett. 1999, 9, 1795–1800. [Google Scholar] [CrossRef]
- Liao, Y; Hendrata, S.; Bae, S-Y.; Wang, W. The effect of phenyl substituents on the release rates of esterase-sensitive coumarin-based prodrugs. Chem. Pharm. Bul. 2000, 48, 1138–1147. [Google Scholar] [CrossRef]
- Achiles, K. Coumarin derivatives as protease-sensitive prodrugs. Arch. Pharm. 2001, 334, 209–215. [Google Scholar] [CrossRef]
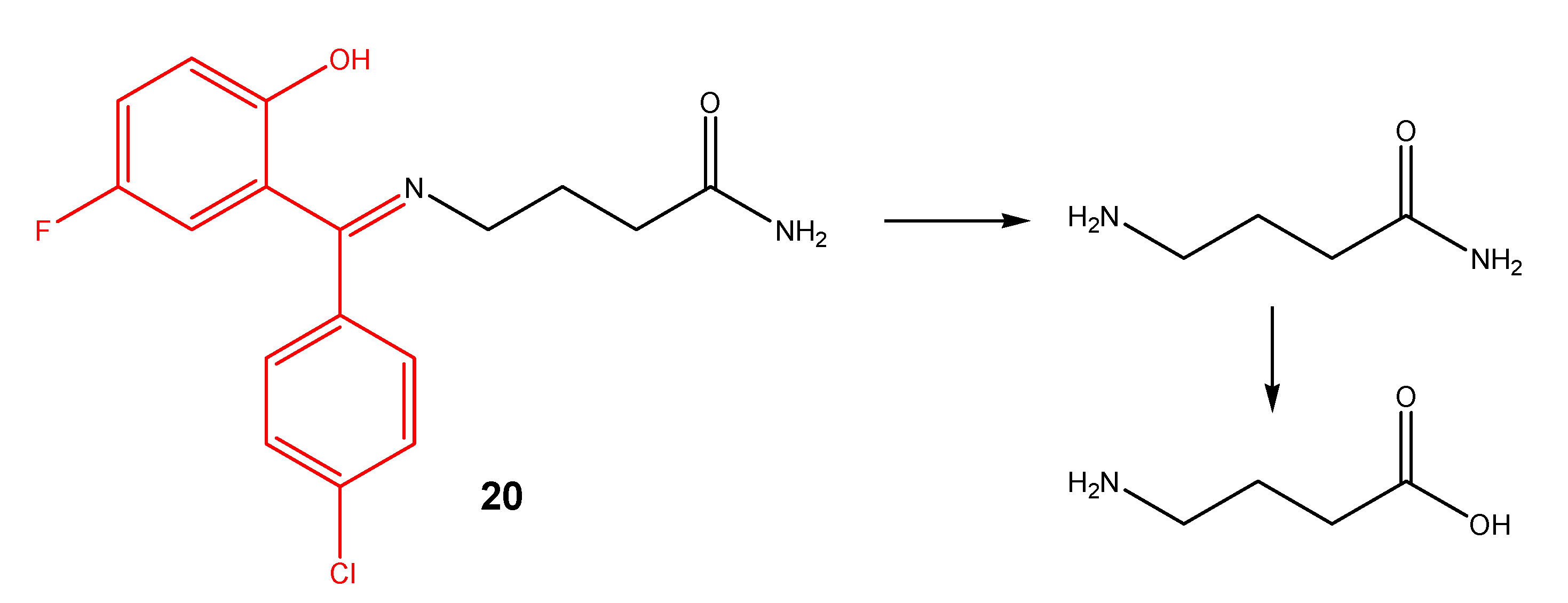
- El-Shorbagi, A. Model for delivery of amines through incorporation into a tetrahydro-2H-1,3,5-thiadiazine-2-thione structure. Eur. J. Med. Chem. 1994, 29, 11–15. [Google Scholar] [CrossRef]
- Aboul-Fadl, T.; El-Shorbagi, A. New prodrug approach for amino acids and amino-acid-like drugs. Eur. J. Med. Chem. 1996, 31, 165–169. [Google Scholar] [CrossRef]
- Aboul-Fadl, T.; El-Shorbagi, A. New carriers for representative peptides and peptide drugs. Arch. Pharm. 1997, 330, 327–332. [Google Scholar] [CrossRef]
- Wang, B.; Wang, W.; Zhang, H.; Shan, D.; Smith, T. Coumarin-based prodrugs. 2. Synthesis and bioreversibility studies of an esterase sensitive cyclic prodrug of Dadle, an opioid peptide. Bioorg. Med. Chem. Lett. 1996, 6, 2823–2826. [Google Scholar] [CrossRef]
- Shan, D.; Nicolaou, M.; Borchardt, R.T.; Wang, B. Prodrug strategies based on intramolecular cyclization reactions. J. Pharm. Sci. 1997, 86, 765–767. [Google Scholar] [CrossRef]
- Pauletti, G.; Gangwar, S.; Wang, B.; Borchardt, R.T. Esterase-sensitive cyclic prodrugs of peptides: evaluation of a phenylpropionic acid promoiety in a model hexapeptide. Pharm. Res. 1997, 14, 11–17. [Google Scholar] [CrossRef]
- Wang, B.; Gangwar, S.; Pauletti, G.; Siahaan, T.; Borchardt, R.T. Synthesis of a novel esterase-sensitive cyclic prodrug system for peptides that utilises a "trimethyl lock"-facilitated lactonization reaction. J. Org. Chem. 1997, 62, 1363–1367. [Google Scholar] [CrossRef]
- Bundgaard, H.; Joahnsen, M. Pro-drugs as drug delivery systems XX. Oxazolidines as potential pro-drug types for β-aminoalcohols, aldehydes and ketones. Int. J. Pharm. 1982, 10, 165–175. [Google Scholar] [CrossRef]
- Rasmussen, G.; Bundgaard, H. Prodrugs of peptides 15. 4-imidazolidinone prodrug derivatives of enkephalins to prevent amino-catalyzed peptidase metabolism in plasma and absortive mucosae. Int. J. Pharm. 1991, 76, 113–122. [Google Scholar] [CrossRef]
- Larsen, S.; Sidenius, M.; Ahkersen, M.; Larsen, C. Kinetics of degradation of 4-imidazolidinone prodrug types obtained from reacting prilocaine with formaldehyde and acetaldehyde. Eur. J. Pharm. Sci. 2003, 20, 233–240. [Google Scholar] [CrossRef]
- Bak, A.; Fich, M.; Larsen, B.; Frokjaer, S.; Friis, G. N-terminal 4-imidazolidinone prodrugs of Leu-enkephalin: synthesis, chemical and enzymatic stability studies. Eur. J. Pharm. Sci. 1999, 7, 317–323. [Google Scholar] [CrossRef]
- Samples Availability: Contact the author.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Simplício, A.L.; Clancy, J.M.; Gilmer, J.F. Prodrugs for Amines. Molecules 2008, 13, 519-547. https://doi.org/10.3390/molecules13030519
Simplício AL, Clancy JM, Gilmer JF. Prodrugs for Amines. Molecules. 2008; 13(3):519-547. https://doi.org/10.3390/molecules13030519
Chicago/Turabian StyleSimplício, Ana L., John M. Clancy, and John F. Gilmer. 2008. "Prodrugs for Amines" Molecules 13, no. 3: 519-547. https://doi.org/10.3390/molecules13030519
APA StyleSimplício, A. L., Clancy, J. M., & Gilmer, J. F. (2008). Prodrugs for Amines. Molecules, 13(3), 519-547. https://doi.org/10.3390/molecules13030519