Introduction
The demand for biodegradable plastics has increased over the past few decades, not only due to increasing environmental concerns, but also for their biomedical applications [
1]. Sucrose is a low molecular weight carbohydrate feedstock from which it is possible to elaborate new materials [
2], such as water-soluble and/or amphiphilic, biocompatible polymers [
3] and other new compounds owing to their low price. Some applications for sugar based polymers are drug delivery systems, dental medicine, bio implants, contact lenses and tissue engineering [
4,
5], and they have the advantage of being potentially biodegradable [
6,
7]. Obtaining polymers and copolymers from carbohydrate derived monomers is a work with a pronounced ecological aspect. The quest for novel, sustainable, but also structurally robust materials is gaining momentum as the pressure on our environment is building up and the progressive changeover of chemical industry to renewable feedstock for their raw materials emerges as an inevitable necessity. To achieve this goal we had proposed the introduction of sugar moieties in the macromolecules of conventional polyolefins by copolymerisation with vinyl carbohydrate derivatives [
8,
9]. Similar types of copolymers, obtained by attaching a sugar moiety to functionalised polyolefins, have been shown to display significantly improved biodegradability [
6,
7].
In previous publications [
8,
9,
10,
11,
12,
13,
14] we have reported the synthesis and applications of different sucrose esters. Since sucrose has eight chemically active hydroxyl groups, regioselective derivatisation is important in the selective synthesis of sucrose-containing linear polymers [
15,
16]. The route to selective derivatisation of the 6'-position of the sucrose has been developed in our laboratory [
12,
13]. It allowed us to obtain the fully protected sucrose with only the 6’-hydroxyl unprotected. Protection-deprotection strategies have been discussed by Jarosz
et al. [
17]. Another difficulty to be taken into account is the instability of the sucrose glycosidic bond under acidic conditions, which prevented us from adapting some efficient catalytic methods allowing the direct coupling of natural polyols with benzyl alcohols over solid acid catalysts, as recently reported [
18].
Herein, we opted to explore the polymerisation activity of π-ether system attached to sucrose and we present the synthesis of novel unsaturated sucrose ethers, as well as an example for their successful free radical copolymerisation with styrene. For obtaining vinyl sucrose ether, we have adopted a new two-step route via mixed acetals, based on the Gassman method, first reported in 1993 [
19], as it requires readily available reagents, mild conditions, and does not involve the use of heavy metal salts catalysts, like mercury [
20]. It consists in forming a vinyl group by the elimination of ethanol from mixed acetals with trimethylsilyl trifluoromethanesulfonate (TMS-triflate) in the presence of alkyl amines. Mixed acetals are readily obtained from the corresponding alcohols and ethyl vinyl ether, using acidic catalysts.
Results and Discussion
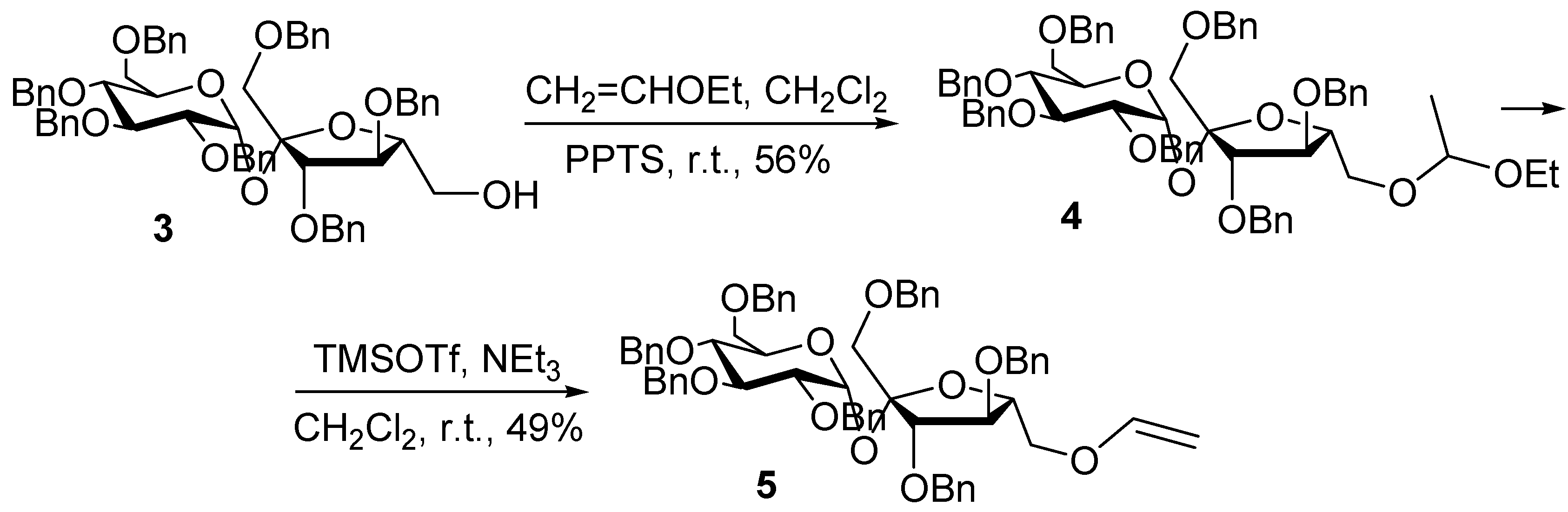
Four unsaturated ether sucrose monomers were synthesised by different methods. The mixed acetal
4 was prepared by treating
3 (obtained as reported [
8]) with ethyl vinyl ether in the presence of PPTS at r.t. for 2 h. No side products were observed, and a mixture of diastereomers was produced, with no attempt at separation being made. After purification by flash column chromatography, the elimination reaction was performed by treating
4 with TMS-triflate and triethylamine at r.t. for 2 h, leading to
5 (
Scheme 1). The yield of this step was decreased by the competing side reaction of formation of silyl ether, resulting of a complexation of the trimethylsilyl cation with the sucrose connected oxygen atom, as it was discussed by Gassman
et al. [
14] and Hughes
et al. [
15]. Complexation of the trimethylsilyl cation with the ethoxy group oxygen produces the desired vinyl sucrose ether.
Scheme 1.
Synthesis of 1’,2,3,3’,4,4’,6-hepta-O-benzyl-6’-O-vinyl sucrose (5).
Scheme 1.
Synthesis of 1’,2,3,3’,4,4’,6-hepta-O-benzyl-6’-O-vinyl sucrose (5).
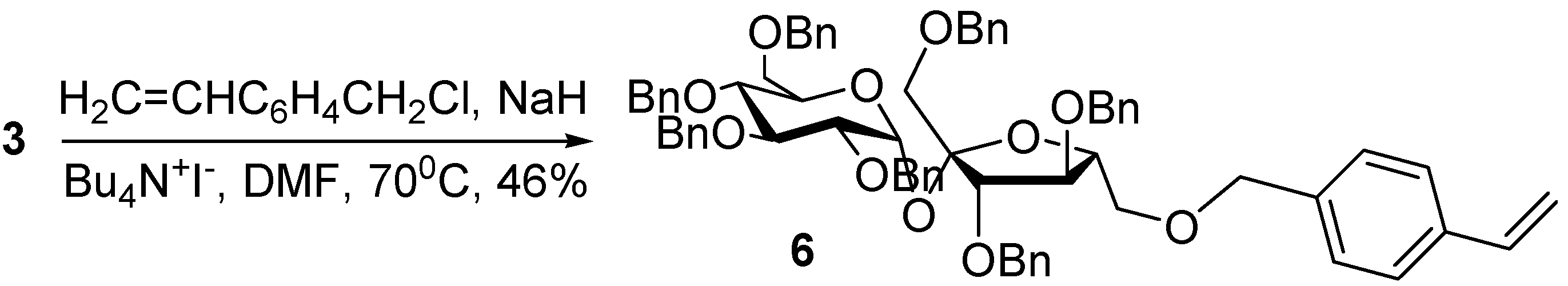
With the purpose of comparing the polymerisation ability of the monomer
5, in which the vinyl group is adjacent to the sucrose moiety, with similar ether compounds in which it is located farther away, we synthesised vinylbenzyl sucrose ethers
6-
8, as previously reported [
3]. We tested conventional etherification involving a primary halide, in our case 4-vinylbenzyl chloride, optimising the benzylation procedure for protected and unprotected sucrose. In the first case, the intermediate
3 was treated with NaH in DMF in the presence of phase-transfer catalyst at 0°C, with the subsequent addition of the 4-vinylbenzyl chloride and increasing the temperature to 70°C (
Scheme 2).
Scheme 2.
Synthesis of 1’,2,3,3’,4,4’,6-hepta-O-benzyl-6’-O-vinylbenzyl sucrose (6).
Scheme 2.
Synthesis of 1’,2,3,3’,4,4’,6-hepta-O-benzyl-6’-O-vinylbenzyl sucrose (6).
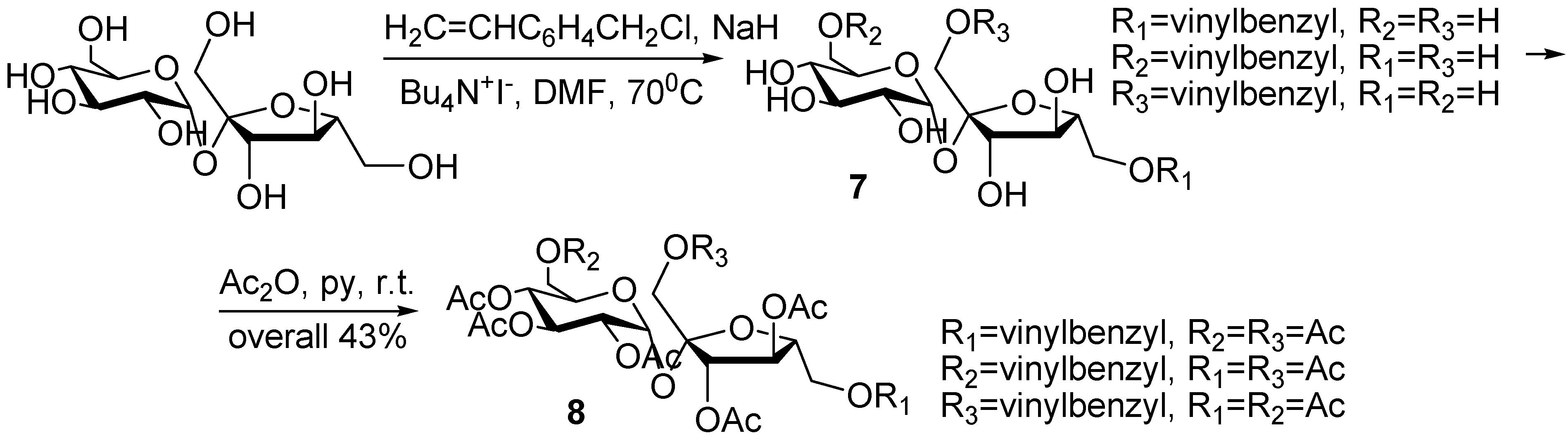
In order to obtain hydrophilic and amphiphilic polymers, we needed monomers with unprotected hydroxyl groups on the sucrose moiety. Those we achieved by reacting sucrose with 4-vinylbenzyl chloride under similar conditions, as described above. The reaction was monitored by TLC, and stopped at the first formation of diethers. A mixture of regioisomers of monovinylbenzyl ethers was isolated, with no attempt at separation being made. Subsequent acetylation was needed for detailed NMR analysis of the reaction products (
Scheme 3).
Scheme 3.
Synthesis of monovinylbenzyl sucrose and hepta-O-acetyl-monovinylbenzyl sucrose (8).
Scheme 3.
Synthesis of monovinylbenzyl sucrose and hepta-O-acetyl-monovinylbenzyl sucrose (8).
The regioselectivity question is a difficult one because of the complex chemistry of sucrose. In general, it is accepted that bulky substituents like TBDPS, are introduced at the primary positions in the order 6-OH ≈ 6’-OH > 1’-OH [
1]. Molecular modeling of sucrose reveals the persistence of an intramolecular hydrogen bond of the 2-O∙∙∙HO-1’ type in aprotic solvents [
21], hence the 2-OH should be the most readily deprotonated under basic conditions, affording the 2-substituted derivative as the major product. In addition, the regioselectivity also depends on the nature of the electrophilic reagent, on the catalyst used for promoting the reaction and, not the least, on the solvent used.
In our case, when we compared the 1H-NMR and COSY spectra of the product and sucrose octa-acetate, the signals for the 2-H had exactly the same chemical shifts, while there were significant differences for the signals of the six methylene protons at the three primary positions. Although this assignment is not precise as we are dealing with a mixture of possible regioisomers, we concluded that most probably the substitution has taken place at the primary positions, the easiest accessible ones, although not the most reactive. The reason for that could be the presence of the phase transfer catalyst Bu4N+I-, or a partial separation of the primary ethers, achieved during the purification in 43% global yield.
With thus obtained monomers example copolymerisation experiments were performed in organic medium, using toluene as a solvent, at 70°C in the presence of AIBN as a radical initiator (
Table 1). The molar mass of the copolymers was measured by dynamic light-scattering, as Mark-Houwink-Sakurada constants were estimated by calibration with commercial polystyrene standards. Copolymer compositions were calculated from
1H NMR spectra by comparing the peak areas of aromatic styrene protons with the 14 sucrose unit protons.
Table 1.
Experimental results for copolymerisation in toluene solutions of 5, 6, and 8 with styrene and the radical initiator AIBN, 70°C, 48 h.
Table 1.
Experimental results for copolymerisation in toluene solutions of 5, 6, and 8 with styrene and the radical initiator AIBN, 70°C, 48 h.
| | [M]0/[S]0[a] | [M]/[S][b] | Yield [%][c] | MW[d] [g/mol] | φ [e] [nm] | α [f] | D[g] [cm2/s] |
|---|
| 5/st | 0.095 | 0.012 | 31 | 31900 | 654 | 0.288 | 5.57.10-9 |
| 6/st | 0.090 | 0.040 | 36 | 6700 | 289 | 0.208 | 1.26.10-8 |
| 8/st | 0.103 | 0.091 | 51 | 89500 | 1121 | 0.251 | 3.25.10-9 |
As we can see, copolymers with good sucrose derivative incorporation rates and high molecular mass were obtained. With monomer
5, in which the double bond is very close to the bulky sucrose moiety and as expected, is impeding the polymerisation, we have lower incorporation of the sugar monomer, but nevertheless high molecular mass for this type of polymers. Under the conditions chosen, we achieved good overall conversions, compared to the reported in the literature [
3], lower incorporation of sugar, which is probably result of the longer polymerisation times, and higher average molecular mass. The optimisation of the polymerisation procedure with a view to possible applications, as well as the biodegradability of copolymers, containing sucrose moieties side groups, is under investigation.
Experimental
General
Reagents and solvents were purified before use [
22]. NMR spectra were recorded at 400 MHz on a Bruker AMX-400 instrument using CDCl
3 as solvent. Chemical shift values (δ) are given in ppm downfield from TMS. The IR spectra were recorded on a Perkin-Elmer FTIR-1600 spectrophotometer. Average molecular weights were determined using a Brookhaven model BI-90 dynamic light-scattering apparatus, with operational angle 90°, calculated from the diffusion coefficients of polymer solutions suspended in ethanol, at 20°C. All solvents used for dilution were filtered through a 0.2 micron membrane filter. 2,500 experimental cycles were used for each run, and the dust cut off, above 21, had no effect on the measurement, indicating a reasonably dust-free sample. The calibration was performed with monodisperse polystyrene standards purchased from Aldrich.
Synthesis of the mixed acetal 1’,2,3,3’,4,4’,6-hepta-O-benzyl-6’-O-(1-ethoxy)ethyl sucrose (4)
To a solution of
3 [
8] (1.016 g; 1.045 mmol) in dry CH
2Cl
2 (10 mL) were added PPTS (10 mg) and ethylvinyl ether (1.5 eq, 0.5 mL). The reaction mixture was stirred at r.t. for 2 h. NaHCO
3 (1 g) was added, and stirred for 10 min more, filtered, concentrated and purified by flash column chromatography on silica gel 60 (0.04-0.06 mm), eluent: hex/EtOAc 3:1. Compound
4 was obtained as a light yellow oil (0.610 g, 56%); Rƒ 0.40 (hex/EtOAc 3:1); IR: ν
max (CH
2Cl
2) 3088, 3040, 3030, 2976, 2897, 2867, 2000-1600, 1496, 1454, 1363, 1266, 1208, 1082, 1028, 736, 698 cm
-1;
1H-NMR: δ
H (CDCl
3) 9.79 (1H, qd
Ј 2.13 Hz, C
H), 7.33-7.20 (32H, m, Ar-H), 7.12-7.10 (2H, m, Ar-H), 5.54 (1H, d
J 2.5 Hz, H-1), 4.92-4.28 (16H, m, C
H2-Ph, H-5’ and H-3), 4.09-4.06 (1H, m, H-6), 4.03-3.97 (2H, m, H-4’ and H-3’), 3.87-3.80 (1H, m, H-6’), 3.73-3.63 (4H, m, H-2, H-1’ and OC
H2CH
3), 3.61-3.51 (4H, m, H-5, H-1’, H-6’ and H-4), 3.48 (1H, d
J 3.03 Hz, H-6), 2.20 (1H, d
J 2.13 Hz, CHC
H3), 1.27-1.19 (4H, m, CH
2C
H3) ppm; confirmed by COSY and HMQC;
13C-NMR: δ
C (CDCl
3) 128.3 (Ph), 128.0 (Ph), 127.9 (Ph), 127.8 (Ph), 127.7 (Ph), 127.5 (Ph), 103.8 (C-2’), 91.1 (C-1), 83.6 (C-5’), 81.8 (C-3’), 81.2 (C-4’), 79.5 (C-3), 79.4 (C-5), 77.3 (C-2), 75.6 (
CH
2-Ph), 74.9 (
CH
2-Ph), 73.5 (
CH
2-Ph), 73.4 (
CH
2-Ph), 73.3 (
CH
2-Ph), 72.9 (
CH
2-Ph), 72.5 (
CH
2-Ph), 71.3 (C-4), 71.2 (C-6’), 67.9 (C-1’), 61.2 (C-6), 60.6 (O
CH
2CH
3), 20.0 (O
2CH
CH3), 15.3 (OCH
2CH
3) ppm; confirmed by DEPT.
1’,2,3,3’,4,4’,6-hepta-O-benzyl-6’-O-vinyl sucrose (5)
To a solution of 4 (0.797 g; 0.763 mmol) in dry CH2Cl2 (7 mL) were added Et3N (0.12 mL, 0.839 mmol) and TMSOTf (0.15 mL, 0.839 mmol) at 0°C. After 10 min it was allowed to warm to r.t. and stirred for 2 h. The reaction mixture was neutralised with 1N NaOH (4 mL), and extracted with diethyl ether (3 x 20 mL). After drying and concentrating, it was purified by flash column chromatography on silica gel 60 (0.04-0.06 mm), eluent: hex/EtOAc 3:1. Compound 5 was obtained as a light yellow oil (0.250 g, 31%); Rƒ 0.5 (Hex/AcOEt 3:1); IR:νmax (CH2Cl2) 3088, 3064, 3030. 2909, 2869, 1737, 2000-1600, 1497, 1454, 1361, 1243, 1209, 1096, 910, 737, 698 cm-1; 1H-NMR: δH (CDCl3) 7.31-7.26 (35H, m, Ar-H), 7.16-7.14 (1H, m, Ar-H), 6.49 (1H, dd Ј 10.73 e 5.1 Hz, OCH=), 5.67 (1H, d J 2.6 Hz, H-1), 4.93 (1H, d J 8.16 Hz, CH2-Ph), 4.83 (1H, d J 8.19 Hz, CH2-Ph), 4.77 (1H, d J 8.16 Hz, CH2-Ph), 4.68-4.38 (12H, m, CH2-Ph and H-5’), 4.20 (1H, dd J 10.8 and 1.41 Hz, =CH2trans), 4.18-4.15 (1H, m, H-3’), 4.10-4.05 (1H, m, H-4), 4.01 (1H, dd J 5.1 and 1.44 Hz, =CH2cis), 3.97-3.92 (1H, m, H-4’), 3.86-3.81 (1H, m, H-6), 3.75 (1H, m, H-6), 3.64 (1H, t J 7.17 Hz, H-2), 3.56-3.49 (3H, m, H-5, H-1’ and H-6’), 3.42 (1H, dd J 7.1 and 1.2 Hz, H-6’) ppm; confirmed by COSY and HMQC; 13C-NMR:δC (CDCl3) 151.6 (OCH=), 128.3 (Ph), 128.3 (Ph), 127.9 (Ph), 127.8 (Ph), 127.7 (Ph), 127.6 (Ph), 127,5 (Ph), 104.6 (C-2’), 90.2 (C-1), 86.9 (=CH2), 83.9 (C-5’), 81.8 (C-3’), 81.2 (C-4’), 79.5 (C-3), 79.4 (C-5), 77.3 (C-2), 75.6 (CH2-Ph), 74.8 (CH2-Ph), 73.4 (CH2-Ph), 72.9 (CH2-Ph), 72.6 (CH2-Ph), 72.3 (CH2-Ph), 71.0 (C-6’), 70.6 (C-4), 69.1 (C-1’), 69.5 (C-6) ppm; confirmed by DEPT.
1’,2,3,3’,4,4’,6-hepta-O-benzyl-6’-O-vinylbenzyl sucrose (6)
To a solution of 3 (0.5 g, 0.514 mmol) in DMF (12.5 mL) were added a catalytic amount of Bu4N+I- and NaH (0.07 g, 1.0 eq) at 0°C. After 20 min, 4-vinylbenzyl chloride (0.09 mL, 1.0 eq) was added, the ice-bath removed, and the reaction mixture was heated at 70°C for 4 h. Concentrated and purified by flash column chromatography on silica gel 60 (0.04-0.06 mm), eluent hex/EtOAc 3:1. Compound 6 was obtained as a light yellow oil (0.260 g, 46%); Rƒ 0.7 (hex/AcOEt 3:1); 1H-NMR: δH (CDCl3) 7.28-7.12 (39H, m, Ar-H), 6.65 (1H, dd Ј 13.2 e 8.2 Hz, PhCH=), 5.71- 5.66 (2H, m, H-1, =CH2trans), 5.20 (1H, dd, J 10.8 e 2.11 Hz =CH2cis), 4.86-4.35 (16 H, m, 7 (CH2-Ph), H-5’, CH2-Ph), 4.22-3.84 (4H, m, H-6’a, H-6’b, H-5, H-4’, H-3), 3.71-3.25 (7H, m, H-2, H-6a, H-6b, H-1’a, H-1’b, H-4, H-3’) ppm; confirmed by COSY and HMQC; 13C-NMR: δC (CDCl3) 138.9 (Ar-C), 138.6 (Ar-C), 138.2 (Ar-C), 138.0 (Ar-C), 137.9 (PhCH=), 136.8 (Ar-C), 136.5 (Ar-C), 128.3 (Ar-CH), 128.3 (Ar-CH), 128.0 (Ar-CH), 127.9 (Ar-CH), 127.7 (Ar-CH), 127.6 (Ar-CH), 127.5 (Ar-CH), 126,2 (Ar), 113.7 (CH2=), 104.6 (C-2’), 89.9 (C-1), 83.9 (C-5’), 82.4 (CH2-Ph), 81.9 (C-3), 81.9 (C-4’), 79.8 (C-3’), 79.6 (C-5), 77.6 (C-2), 75.5 (CH2-Ph), 74.8 (CH2-Ph), 73.4 (CH2-Ph), 73.0 (CH2-Ph), 72.5 (CH2-Ph), 72.2 (CH2-Ph), 71.4 (C-6’), 71.2 (C-6), 70.6 (C-4), 68.4 (C-1’) ppm; confirmed by DEPT.
Hepta-O-acetyl-monovinylbenzyl sucrose (8).
To a solution of sucrose (1.000 g, 2.9 mmol) in DMF (25 mL) were added a catalytic amount of Bu4N+I- and NaH (0.14 g, 1.1 eq) at 0°C. After 20 min, 4-vinylbenzyl chloride (0.45 ml, 1.1 eq) was added, the ice-bath removed, and the reaction mixture was heated at 70°C for 4 h. After removing the solvent, the residue was dissolved in pyridine, acetic anhydride (3 mL) was added, and the mixture stirred overnight. Concentrated and purified by flash column chromatography on silica gel 60 (0.04-0.06 mm), eluent hex/EtOAc 1:1. Compound 8 was obtained as a light yellow oil (0.934 g, 43%); Rƒ 0.7 (hex/AcOEt 1:2); IR:νmax (CH2Cl2) 3059, 2962, 2121, 1746, 1513, 1432, 1370, 1230, 1168, 1104, 1046, 910, 829, 737, 703, 601 cm-1; 1H-NMR: δH (CDCl3) 7.56-7.43 (2H, m, Ar-H), 7.27-7.21 (2H, m, Ar-H), 6.71 (1H, dd Ј 13.2 e 8.2 Hz, PhCH=), 5.75 (1H, dd J 13.2 e 4.02 Hz, =CH2trans), 5.72-5.65 (1H, m, H-1), 5.56-5.5 (1H, m, H-4’), 5.42 (1H, t Ј 6.56 Hz, H-3), 5.40-5.36 (1H, m, H-3), 5.30 (1H, s, PhCH2), 5.27 (1H, dd J 9.14 e 3.48 Hz, =CH2cis), 5.05 (1H, m, H-4), 4.88-4.81 (1H, dd, H-2), 4.66-4.45 (2H, m, H-6’a, H-6’b ), 4.38-3.97 (7H, m, H-1’a, H-1’b, H-6a, H-6b, H-5, H-5’), 3.61-3.5 (1H, m), 2.16-1.90 (21H, m, CH3(C=O)) ppm; confirmed by COSY and HMQC; 13C-NMR: δC (CDCl3) 170.7-169.8 (CH3(C=O)), 137.5 (Ar), 137.3 (Ar), 137.0 (Ar), 136.7 (Ar), 136.4 (PhCH=), 128.8 (Ar), 128.5 (Ar), 128.3 (Ar), 128.0 (Ar), 126.4 (Ar), 126.3 (Ar), 114.0 (CH2=), 113.8 (CH2=), 104.5 (C-2’), 103.6 (C-2’), 90.3 (C-1), 89.6 (CH2-Ph), 78.8 (C-5’), 78.5 (C-5’), 75.8 (C-3), 75.4 (C-4’), 74.6 (C-3’), 73.4 (C-6’), 72.5 (C-6), 71.7 (C-2), 70.2 (C-1’), 68.5-68.3 (C-4 and C-5), 63.7-63.3 (C-6 and C-6’), 61.9 (C-1’), 61.7 (C-1’), 20.7 (CH3(C=O)) ppm; confirmed by DEPT.
General procedure for free radical copolymerisations of monomers 5, 6, and 8 with styrene
Copolymerisations were carried out in anhyd. toluene solutions (0.1 M) in the presence of AIBN as radical initiator (1 % by weight with respect to the monomer mixture). Dissolved oxygen was removed from the solutions by sparging with argon. They were then heated at 70°C for 48 h, and the solutions were then cooled to r.t. and the product precipitated in cold EtOH. The white solid was filtered and washed several times with cold EtOH. The polymers were purified by repeated dissolution in toluene and reprecipitation in cold EtOH and dried under vacuum.





{kind=link}
{kind=link}
{kind=link}