Enhanced Absorption and Growth Inhibition with Amino Acid Monoester Prodrugs of Floxuridine by Targeting hPEPT1 Transporters

Abstract
:Introduction
Results and Discussion


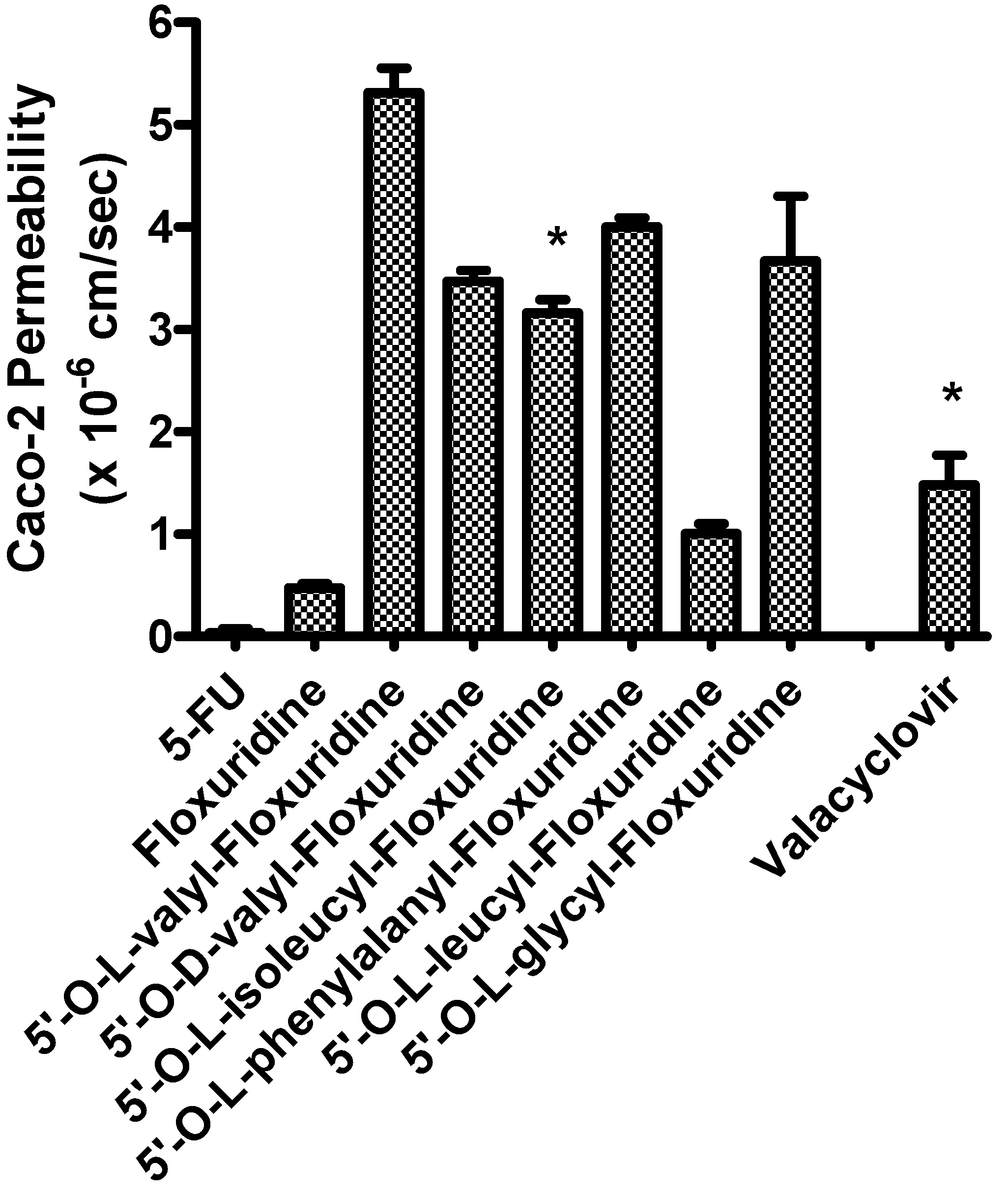{kind=link}
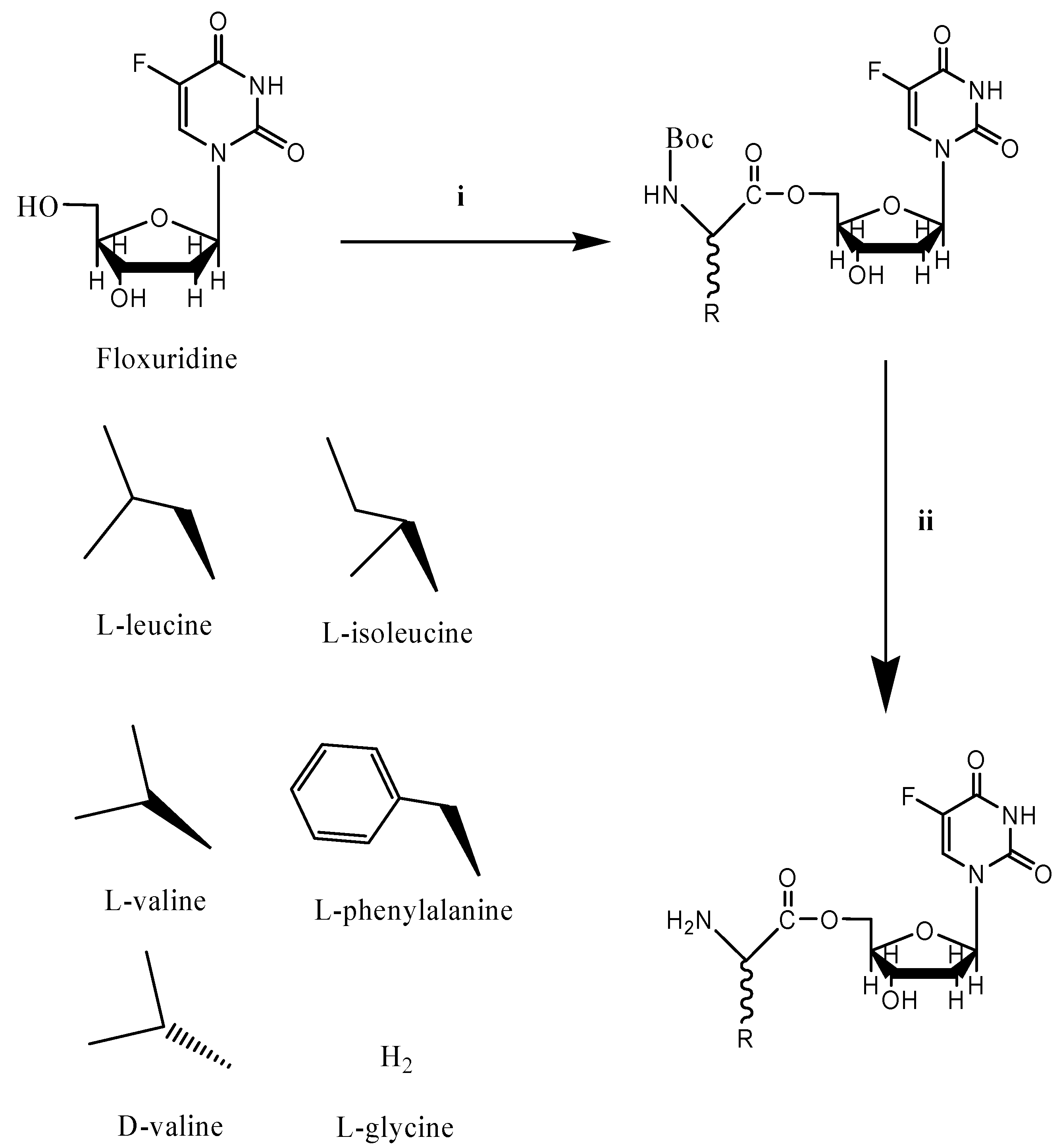{kind=link}
| Required | Observed | ||||
|---|---|---|---|---|---|
| 5'-O-L-leucyl-floxuridine | 96.77 | 360.2 | 360.4 | 473.4 | -0.95 |
| 5'-O-L-phenylalanyl-floxuridine | 96.10 | 394.2 | 394.0 | 507.4 | -0.51 |
| 5'-O-L-valyl-floxuridine | 98.51 | 346.2 | 346.0 | 459.4 | -1.30 |
| 5'-O-D-valyl-floxuridine | 99.52 | 346.2 | 346.0 | 459.4 | -1.30 |
| 5'-O-L-isoleucyl-floxuridine | 95.96 | 360.2 | 360.4 | 473.4 | -0.78 |
| 5'-O-L-glycyl-floxuridine | 95.23 | 304.2 | 303.9 | 417.4 | -2.68 |
| Half Life (min) | ||||
|---|---|---|---|---|
| Prodrug | Buffer pH 7.4 | Homogenates from Caco-2 cells | Homogenates from AsPC-1 cells | Homogenates from MDCK cells |
| Floxuridine | nd | 5.7 ± 0.3 | 6.4 ± 3.2 | 68.9 ± 12.8 |
| 5'-O-l-valyl-floxuridine | 303.9 ± 17.8 | 9.4 ± 0.6 | 18.7 ± 6.7 | 74.7 ± 5.3 |
| 5'-O-d-valyl-floxuridine | 344.9 ± 10.2 | 342.6 ± 120.2 | 290.9 ± 48.9 | 311.6 ± 45.4 |
| 5'-O-l-phenylalanyl-floxuridine | 221.7 ± 56.7 | 11.1 ± 9.9 | 11.8 ± 1.7 | 6.0 ± 0.6 |
| 5'-O-l-leucyl-floxuridine | 77.3 ± 1.2 | 4.8 ± 0.2 | 2.0 ± 0.1 | 9.2 ± 1.1 |
| 5'-O-l-isoleucyl-floxuridine | 323.5 ± 1.5a | 192.3 ± 31.8 | 198.0 ± 34.1 | 244.9 ± 18.3 |
| 5'-O-l-glycyl-floxuridine | 85.5 ± 3.2 | 24.1 ± 2.0 | 27.6 ± 5.8 | 11.2 ± 2.0 |
| Prodrug | Caco-2 cell | AsPC-1 cell |
|---|---|---|
| Floxuridine | 7.3 ± 1.6 | 6.3 ± 2.3 |
| 5'-O-l-valyl-floxuridine | 1.0 ± 0.1 | 2.9 ± 0.4 |
| 5'-O-d-valyl-floxuridine | 2.6 ± 0.1 | 4.8 ± 1.3 |
| 5'-O-l-phenylalanyl-floxuridine | 2.1 ± 0.1 | 2.0 ± 0.1 |
| 5'-O-l-leucyl-floxuridine | 2.0 ± 0.3 | 2.6 ± 0.2 |
| 5'-O-l-isoleucyl-floxuridine | 0.7 ± 0.0 | 4.1 ± 1.8 |
| 5'-O-l-glycyl-floxuridine | 2.3 ± 0.6 | 2.7 ± 0.5 |

| GI50 (µM) | |||
|---|---|---|---|
| Prodrug | MDCK | MDCK/hPEPT1 | Enhancement Factor |
| 5'-O- l-valyl-floxuridine | 126.6 ± 7.7 | 21.1 ± 4.2 | 5.91 |
| 5'-O- d-valyl-floxuridine | 88.5 ± 2.6 | 71.0 ± 2.7 | 1.25 |
| 5'-O- l-phenylalanyl-floxuridine | 41.3 ± 3.9 | 4.7 ± 2.4 | 8.80 |
| 5'-O- l-leucyl-floxuridine | 23.5 ± 3.0 | 2.3 ± 1.0 | 10.09 |
| 5'-O- l-isoleucyl-floxuridine | 186.5 ± 4.2 | 10.3 ± 2.9 | 18.04 |
| Values are presented as mean ± S.D.; The prodrug concentration required to inhibit growth by 50 % (GI50) was determined in MDCK cells and MDCK cells that overexpressed hPEPT1 (MDCK/hPEPT1). The ratios of GI50 in MDCK and MDCK/hPEPT1 cells are presented as Enhancement Factor. | |||
Conclusions
Experimental
Materials
Floxuridine Prodrug Synthesis
Cell Culture
Hydrolysis Studies
Data Analysis

HPLC Analysis
[3H]Gly-Sar Uptake Inhibition
Caco-2 permeability study
Cell Proliferation Assays
Acknowledgements
References and Notes
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer, R.J.; Pleven, E.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–6. [Google Scholar] [CrossRef]
- Cohen, AM.; Minsky, B. D.; Schilsky, R. L. Cancer, Principles and Practices in Oncology, 4 Edition ed; J.B. Lippincott co.: Philadelphia, 1993; pp. 929–71. [Google Scholar]
- Grem, J.L. 5-Fluorouracil: forty-plus and still ticking. A review of its preclinical and clinical development. Invest. New Drugs 2000, 18, 299–313. [Google Scholar] [CrossRef]
- Willmore, E.; Durkacz, B.W. Cytotoxic mechanisms of 5-fluoropyrimidines. Relationships with poly(ADP-ribose) polymerase activity, DNA strand breakage and incorporation into nucleic acids. Biochem. Pharmacol. 1993, 46, 205–11. [Google Scholar] [CrossRef]
- Parker, W.B.; Cheng, Y.C. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol. Ther. 1990, 48, 381–95. [Google Scholar] [CrossRef]
- van Laar, J.A.; Rustum, Y.M.; Ackland, S.P.; van Groeningen, C.J.; Peters, G.J. Comparison of 5-fluoro-2'-deoxyuridine with 5-fluorouracil and their role in the treatment of colorectal cancer. Eur. J. Cancer. 1998, 34, 296–306. [Google Scholar]
- Shibamoto, Y.; Tachi, Y.; Tanabe, K.; Hatta, H.; Nishimoto, S. In vitro and in vivo evaluation of novel antitumor prodrugs of 5-fluoro-2'-deoxyuridine activated by hypoxic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 397–402. [Google Scholar] [CrossRef]
- Yamada, M.; Nakagawa, H.; Fukushima, M.; Shimizu, K.; Hayakawa T, Ikenaka K. In vitro study on intrathecal use of 5-fluoro-2'-deoxyuridine (FdUrd) for meningeal dissemination of malignant brain tumors. J. Neurooncol. 1998, 37, 115–21. [Google Scholar]
- Laskin, J.D.; Evans, R.M.; Slocum, H.K.; Burke, D.; Hakala, M.T. Basis for natural variation in sensitivity to 5-fluorouracil in mouse and human cells in culture. Cancer. Res. 1979, 39, 383–90. [Google Scholar]
- Birnie, G.D.; Kroeger, H.; Heidelberger, C. Studies Of Fluorinated Pyrimidines. Xviii. The Degradation Of 5-Fluoro-2'-Deoxyuridine And Related Compounds By Nucleoside Phosphorylase. Biochemistry 1963, 13, 566–72. [Google Scholar]
- Kim, I.; Chu, X.Y.; Kim, S.; Provoda, C.J.; Lee, K.D.; Amidon, G.L. Identification of a human valacyclovirase: biphenyl hydrolase-like protein as valacyclovir hydrolase. J. Biol. Chem. 2003, 278, 25348–56. [Google Scholar]
- Kim, I.; Song, X.; Vig, B.S.; Mittal, S.; Shin, H.C.; Lorenzi, P.J.; Amidon, G.L. A novel nucleoside prodrug-activating enzyme: substrate specificity of biphenyl hydrolase-like protein. Mol. Pharm. 2004, 1, 117–27. [Google Scholar] [CrossRef]
- Landowski, C.P.; Lorenzi, P.L.; Song, X.; Amidon, G.L. Nucleoside ester prodrug substrate specificity of liver carboxylesterase. J. Pharmacol. Exp. Ther. 2006, 316, 572–80. [Google Scholar]
- Han, H.K.; Oh, D.M.; Amidon, G.L. Cellular uptake mechanism of amino acid ester prodrugs in Caco-2/hPEPT1 cells overexpressing a human peptide transporter. Pharm. Res. 1998, 15, 1382–6. [Google Scholar] [CrossRef]
- Landowski, C.P.; Vig, B.S.; Song, X.; Amidon, G.L. Targeted delivery to PEPT1-overexpressing cells, acidic, basic, and secondary floxuridine amino acid ester prodrugs. Mol. Cancer Ther. 2005, 4, 659–67. [Google Scholar] [CrossRef]
- Anand, B.S.; Katragadda, S.; Mitra, A.K. Pharmacokinetics of novel dipeptide ester prodrugs of acyclovir after oral administration: intestinal absorption and liver metabolism. J. Pharmacol. Exp. Ther. 2004, 311, 659–67. [Google Scholar] [CrossRef]
- Anand, B.S.; Patel, J.; Mitra, A.K. Interactions of the dipeptide ester prodrugs of acyclovir with the intestinal oligopeptide transporter: competitive inhibition of glycylsarcosine transport in human intestinal cell line-Caco-2. J. Pharmacol. Exp. Ther. 2003, 304, 781–91. [Google Scholar] [CrossRef] [Green Version]
- Meredith, D.; Temple, C.S.; Guha, N.; Sword, C.J.; Boyd, C.A.; Collier, I.D.; Morgan, K.M.; Bailey, P.D. Modified amino acids and peptides as substrates for the intestinal peptide transporter PepT1. Eur. J. Biochem. 2000, 267, 3723–8. [Google Scholar] [CrossRef]
- Surendran, N.; Covitz, K.M.; Han, H.; Sadee, W.; Oh, D.M.; Amidon, G.L.; Williamson, R.M.; Bigge, C.F.; Stewart, B.H. Evidence for overlapping substrate specificity between large neutral amino acid (LNAA) and dipeptide (hPEPT1) transporters for PD 158473, an NMDA antagonist. Pharm. Res. 1999, 16, 391–5. [Google Scholar] [CrossRef]
- Wenzel, U.; Thwaites, D.T.; Daniel, H. Stereoselective uptake of beta-lactam antibiotics by the intestinal peptide transporter. Br. J. Pharmacol. 1995, 116, 3021–7. [Google Scholar] [CrossRef]
- Nielsen, C.U.; Andersen, R.; Brodin, B.; Frokjaer, S.; Taub, M.E.; Steffansen, B. Dipeptide model prodrugs for the intestinal oligopeptide transporter. Affinity for and transport via hPepT1 in the human intestinal Caco-2 cell line. J. Control. Release. 2001, 76, 129–38. [Google Scholar] [CrossRef]
- Satake, M.; Enjoh, M.; Nakamura, Y.; Takano, T.; Kawamura, Y.; Arai, S.; Shimizu, M. Transepithelial transport of the bioactive tripeptide, Val-Pro-Pro, in human intestinal Caco-2 cell monolayers. Biosci. Biotechnol. Biochem. 2002, 66, 378–84. [Google Scholar] [CrossRef]
- Wenzel, U.; Gebert, I.; Weintraut, H.; Weber, W.M.; Clauss, W.; Daniel, H. Transport characteristics of differently charged cephalosporin antibiotics in oocytes expressing the cloned intestinal peptide transporter PepT1 and in human intestinal Caco-2 cells. J. Pharmacol. Exp. Ther. 1996, 277, 831–9. [Google Scholar]
- Han, H.; de Vrueh, R.L.; Rhie, J.K.; Covitz, K.M.; Smith, P.L.; Lee, C.P.; Oh, D.M.; Sadee, W.; Amidon, G.L. 5'-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm. Res. 1998, 15, 1154–9. [Google Scholar] [CrossRef]
- Weller, S.; Blum, M.R.; Doucette, M.; Burnette, T.; Cederberg, D.M.; de Miranda, P.; Smiley, M.L. Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers. Clin. Pharmacol. Ther. 1993, 54, 595–605. [Google Scholar] [CrossRef]
- Gonzalez, D.E.; Covitz, K.M.; Sadee, W.; Mrsny, R.J. An oligopeptide transporter is expressed at high levels in the pancreatic carcinoma cell lines AsPc-1 and Capan-2. Cancer Res. 1998, 58, 519–25. [Google Scholar]
- Nakanishi, T.; Tamai, I.; Takaki, A.; Tsuji, A. Cancer cell-targeted drug delivery utilizing oligopeptide transport activity. Int. J. Cancer. 2000, 88, 274–80. [Google Scholar] [CrossRef]
- Friedrichsen, G.M.; Chen, W.; Begtrup, M.; Lee, C.P.; Smith, P.L.; Borchardt, R.T. Synthesis of analogs of L-valacyclovir and determination of their substrate activity for the oligopeptide transporter in Caco-2 cells. Eur. J. Pharm. Sci. 2002, 16, 1–13. [Google Scholar] [CrossRef]
- Guo, A.; Hu, P.; Balimane, P.V.; Leibach, F.H.; Sinko, P.J. Interactions of a nonpeptidic drug, valacyclovir, with the human intestinal peptide transporter (hPEPT1) expressed in a mammalian cell line. J. Pharmacol. Exp. Ther. 1999, 289, 448–54. [Google Scholar]
- Landowski, C.P.; Sun, D.; Foster, D.R.; Menon, S.S.; Barnett, J.L.; Welage, L.S.; Ramachandran, C.; Amidon, G.L. Gene expression in the human intestine and correlation with oral valacyclovir pharmacokinetic parameters. J. Pharmacol. Exp. Ther. 2003, 306, 778–86. [Google Scholar] [CrossRef]
- Phan, D.D.; Chin-Hong, P.; Lin, E.T.; Anderle, P.; Sadee, W.; Guglielmo, B.J. Intra- and interindividual variabilities of valacyclovir oral bioavailability and effect of coadministration of an hPEPT1 inhibitor. Antimicrob. Agents Chemother. 2003, 47, 2351–3. [Google Scholar]
- Umapathy, N.S.; Ganapathy, V.; Ganapathy, M.E. Transport of amino acid esters and the amino-acid-based prodrug valganciclovir by the amino acid transporter ATB(0,+). Pharm. Res. 2004, 21, 1303–10. [Google Scholar] [CrossRef]
- Hu, M.; Subramanian, P.; Mosberg, H.I.; Amidon, G.L. Use of the peptide carrier system to improve the intestinal absorption of L-alpha-methyldopa: carrier kinetics, intestinal permeabilities, and in vitro hydrolysis of dipeptidyl derivatives of L-alpha-methyldopa. Pharm. Res. 1989, 6, 66–70. [Google Scholar] [CrossRef]
- Anand, B.S.; Dey, S.; Mitra, A.K. Current prodrug strategies via membrane transporters/receptors. Expert. Opin. Biol. Ther. 2002, 2, 607–20. [Google Scholar] [CrossRef]
- Ganapathy, M.E.; Huang, W.; Wang, H.; Ganapathy, V.; Leibach, F.H. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem. Biophys. Res. Commun. 1998, 246, 470–5. [Google Scholar] [CrossRef]
- Eriksson, A.H.; Elm, P.L.; Begtrup, M.; Nielsen, R.; Steffansen, B.; Brodin, B. hPEPT1 affinity and translocation of selected Gln-Sar and Glu-Sar dipeptide derivatives. Mol. Pharm. 2005, 2, 242–9. [Google Scholar] [CrossRef]
- Li, J.; Tamura, K.; Lee, C.P.; Smith, P.L.; Borchardt, R.T.; Hidalgo, I.J. Structure-affinity relationships of Val-Val and Val-Val-Val stereoisomers with the apical oligopeptide transporter in human intestinal Caco-2 cells. J. Drug. Target. 1998, 5, 317–27. [Google Scholar] [CrossRef]
- Tamura, K.; Bhatnagar, P.K.; Takata, J.S.; Lee, C.P.; Smith, P.L.; Borchardt, R.T. Metabolism, uptake, and transepithelial transport of the diastereomers of Val-Val in the human intestinal cell line, Caco-2. Pharm. Res. 1996, 13, 1213–8. [Google Scholar] [CrossRef]
- Vabeno, J.; Lejon, T.; Nielsen, C.U.; Steffansen, B.; Chen, W.; Ouyang, H.; Borchardt, R.T. Phe-Gly dipeptidomimetics designed for the di-/tripeptide transporters PEPT1 and PEPT2: synthesis and biological investigations. J. Med. Chem. 2004, 47, 1060–9. [Google Scholar] [CrossRef]
- Landowski, C.P.; Song, X.; Lorenzi, P.L.; Hilfinger, J.M.; Amidon, G.L. Floxuridine amino Acid ester prodrugs: enhancing Caco-2 permeability and resistance to glycosidic bond metabolism. Pharm. Res. 2005, 22, 1510–8. [Google Scholar]
- Lorenzi, P.L.; Landowski, C.P.; Song, X.; Borysko, K.Z.; Breitenbach, J.M.; Kim, J.S.; Hilfinger, J.M.; Townsend, L.B.; Drach, J.C.; Amidon, G.L. Amino acid ester prodrugs of 2-bromo-5,6-dichloro-1-(beta-D-ribofuranosyl)benzimidazole enhance metabolic stability in vitro and in vivo. J. Pharmacol. Exp. Ther. 2005, 314, 883–90. [Google Scholar] [CrossRef]
- Song, X.; Vig, B.S.; Lorenzi, P.L.; Drach, J.C.; Townsend, L.B.; Amidon, G.L. Amino acid ester prodrugs of the antiviral agent 2-bromo-5,6-dichloro-1-(beta-D-ribofuranosyl)benzimidazole as potential substrates of hPEPT1 transporter. J. Med. Chem. 2005, 48, 1274–7. [Google Scholar] [CrossRef]
- Vig, B.S.; Lorenzi, P.J.; Mittal, S.; Landowski, C.P.; Shin, H.C.; Mosberg, H.I.; Hilfinger, J.M.; Amidon, G.L. Amino acid ester prodrugs of floxuridine: synthesis and effects of structure, stereochemistry, and site of esterification on the rate of hydrolysis. Pharm. Res. 2003, 20, 1381–8. [Google Scholar] [CrossRef]
- Daniel, H.; Morse, E.L.; Adibi, S.A. Determinants of substrate affinity for the oligopeptide/H+ symporter in the renal brush border membrane. J. Biol. Chem. 1992, 267, 9565–73. [Google Scholar]
- Fang, G.; Konings, W.N.; Poolman, B. Kinetics and substrate specificity of membrane-reconstituted peptide transporter DtpT of Lactococcus lactis. J. Bacteriol. 2000, 182, 2530–5. [Google Scholar]
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsume, Y.; Vig, B.S.; Sun, J.; Landowski, C.P.; Hilfinger, J.M.; Ramachandran, C.; Amidon, G.L. Enhanced Absorption and Growth Inhibition with Amino Acid Monoester Prodrugs of Floxuridine by Targeting hPEPT1 Transporters. Molecules 2008, 13, 1441-1454. https://doi.org/10.3390/molecules13071441
Tsume Y, Vig BS, Sun J, Landowski CP, Hilfinger JM, Ramachandran C, Amidon GL. Enhanced Absorption and Growth Inhibition with Amino Acid Monoester Prodrugs of Floxuridine by Targeting hPEPT1 Transporters. Molecules. 2008; 13(7):1441-1454. https://doi.org/10.3390/molecules13071441
Chicago/Turabian StyleTsume, Yasuhiro, Balvinder S. Vig, Jing Sun, Christopher P. Landowski, John M. Hilfinger, Chandrasekharan Ramachandran, and Gordon L. Amidon. 2008. "Enhanced Absorption and Growth Inhibition with Amino Acid Monoester Prodrugs of Floxuridine by Targeting hPEPT1 Transporters" Molecules 13, no. 7: 1441-1454. https://doi.org/10.3390/molecules13071441
APA StyleTsume, Y., Vig, B. S., Sun, J., Landowski, C. P., Hilfinger, J. M., Ramachandran, C., & Amidon, G. L. (2008). Enhanced Absorption and Growth Inhibition with Amino Acid Monoester Prodrugs of Floxuridine by Targeting hPEPT1 Transporters. Molecules, 13(7), 1441-1454. https://doi.org/10.3390/molecules13071441