Microwave Synthesis of Quaternary Ammonium Salts

Abstract
:Introduction

Results and Discussion


{kind=link}
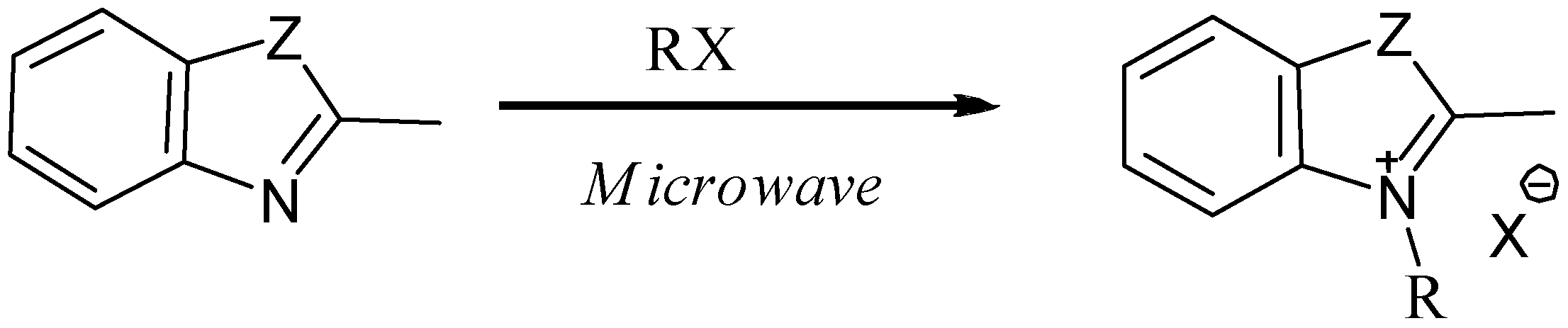{kind=link}
| Entry | Z | R | Temp (οC) | Time (min) | Yield (%) | Lit. Yield (%) | Lit Time (h) |
|---|---|---|---|---|---|---|---|
| 1 | C(CH3)2 | Et | 130 | 5:00 | 95 | 59 | 48 [13] |
| 2 | C(CH3)2 | Me | 110 | 2:30 | 93 | 75 | 21 [11] |
| 3 | C(CH3)2 | Pr | 110 | 7:00 | 83 | 44 | 24 [15] |
| 4 | C(CH3)2 | -(CH2)2OH | 110 | 7:00 | 73 | 69 | 24 [16] |
| 5 | C(CH3)2 | -(CH2)5CO2H | 110 | 7:00 | 59 | 67 | 12 [15] |
| 6 | S | Et | 170 | 20:00 | 83 | 48 | 48 [13] |
| 7 | S | Me | 120 | 20:00 | 85 | 60 | 7 [17] |
| 8 | S | Pr | 170 | 20:00 | 65 | 5 | 7 [17] |
| 9 | S | -(CH2)2OH | 100 | 35:00 | 58 | N/A | 6 [18] |
| 10 | S | -(CH2)5CO2H | 170 | 35:00 | 51 | 61 | 48 [19] |
Work up and purification
Conclusions
Experimental
General
Acknowledgements
References and Notes
- Zhu, H.; Clark, S. M.; Benson, S. C.; Rye, H. S.; Glazer, A. N.; Mathies, R. A. High-sensitivity capillary electrophoresis of double-stranded DNA fragments using monomeric and dimeric fluorescent intercalating dyes. Anal. Chem. 1994, 66, 1941–1948. [Google Scholar] [CrossRef]
- Schwartz, H. E.; Ulfelder, K. J. Capillary electrophoresis with laser-induced fluorescence detection of PCR fragments using thiazole orange. Anal. Chem. 1992, 64, 1737–1740. [Google Scholar] [CrossRef]
- Bengtsson, M.; Karlsson, H. J.; Westman, G.; Kubista, M. A new minor groove binding asymmetric cyanine reporter dye for real-time PCR. Nucleic Acids Res. 2003, 31, e45/1. [Google Scholar]
- Hirons, G. T.; Fawcett, J. J.; Crissman, H. A. TOTO and YOYO: New very bright fluorochromes for DNA content analyses by flow cytometry. Cytometry 1994, 15, 129–140. [Google Scholar] [CrossRef]
- Kurihara, K.; Toyoshima, Y.; Sukigara, M. Phase transition and dye aggregation in phospholipid-amphiphilic dye liposome bilayers. J. Phys. Chem. 1977, 81, 1833–1837. [Google Scholar] [CrossRef]
- Armitage, B.; O'Brien, D. F. Vectorial photoinduced electron transfer between phospholipid membrane-bound donors and acceptors. J. Am. Chem. Soc. 1992, 114, 7396–7403. [Google Scholar] [CrossRef]
- Reers, M.; Smith, T. W.; Chen, L. B. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 1991, 30, 4480–4486. [Google Scholar] [CrossRef]
- Pawley, J. B. (Ed.) Handbook of Confocal Microscopy, 2nd ed.; Plenum Press: New York, USA, 1995.
- Mirshra, A.; Behera, R.K.; Behera, B.K.; Mishra, B.K.; Behera, G.B. Cyanines during the 1990s: A Review. Chem. Rev. 2000, 100, 1973–2011. [Google Scholar] [CrossRef]
- Hirano, M.; Osakada, K.; Nohira, H.; Miyashita, A. Crystal and solution structures of photochromic spirobenzothiopyran. First full characterization of the meta-stable colored species. J. Org. Chem. 2002, 67, 533–540. [Google Scholar] [CrossRef]
- Narayanan, N.; Patonay, G. A New Method for the Synthesis of heptamethine cyanine dyes: synthesis of new near-infrared fluorescent labels. J. Org. Chem. 1995, 60, 2391–2395. [Google Scholar] [CrossRef]
- Pardal, A.C.; Ramos, S.S.; Santos, P.F.; Reis, L.V.; and Almeida, P. Synthesis and spectroscopic characterisation of n-Alkyl quaternary ammonium salts typical precursors of cyanines. Molecules 2002, 7, 320–330. [Google Scholar] [CrossRef]
- Elizalde, L.E.; Ledezma, R.; Lopez, R.G. Synthesis of Potochromic Monomers derived form 1’-(2-Methacryloxyethyl)-3,3-Dimethyl-2-[2H]-Spirobenzopyran Indoline. Synth. Commun. 2005, 35, 603–610. [Google Scholar]
- Winstead, A.J.; Williams, R.; Hart, K.; Fleming, N.; Kennedy, A. Microwave synthesis of near infrared heptamethine cyanine dye. J. Microw. Power Electromagn. Energy 2008, 42, 35–41. [Google Scholar]
- Jung, M.E.; Kim, W. Practical synthesis of dyes for difference gel electrophoresis. Bio. Med. Chem. 2006, 14, 92–97. [Google Scholar] [CrossRef]
- Raymo, F.M.; Giordani, S. Signal processing at the molecular level. J. Am. Chem. Soc. 2001, 123, 4651–4652. [Google Scholar] [CrossRef]
- Kuramoto, N.; Natsukawa, K.; Asao, K. Synthesis and characterization of deep-coloured squarylium dyes for laser optical recording media. Dyes Pigments 1989, 11, 21–35. [Google Scholar] [CrossRef]
- Ye, C.; Ren, J.; Ge, H.; Lu, X. Synthesis of 2-substituted benzothiazoline derivatives. Hecheng Huaxue 2005, 13, 206–207. [Google Scholar]
- Ye, C.; Ren, J.; Ge, H.; Lu, X. Syntheses of novel p-phenylenediethenylenedicyanine dyes. Huaxue Yanjiu Yu Yingyong 2004, 16, 635–638. [Google Scholar]
- Sample Availability: Samples of compounds 1-10 are available from the authors
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Winstead, A.J.; Fleming, N.; Hart, K.; Toney, D. Microwave Synthesis of Quaternary Ammonium Salts. Molecules 2008, 13, 2107-2113. https://doi.org/10.3390/molecules13092107
Winstead AJ, Fleming N, Hart K, Toney D. Microwave Synthesis of Quaternary Ammonium Salts. Molecules. 2008; 13(9):2107-2113. https://doi.org/10.3390/molecules13092107
Chicago/Turabian StyleWinstead, Angela J., Nicole Fleming, Krystal Hart, and Deveine Toney. 2008. "Microwave Synthesis of Quaternary Ammonium Salts" Molecules 13, no. 9: 2107-2113. https://doi.org/10.3390/molecules13092107
APA StyleWinstead, A. J., Fleming, N., Hart, K., & Toney, D. (2008). Microwave Synthesis of Quaternary Ammonium Salts. Molecules, 13(9), 2107-2113. https://doi.org/10.3390/molecules13092107