Ring-substituted 4-Hydroxy-1H-quinolin-2-ones: Preparation and Biological Activity



 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
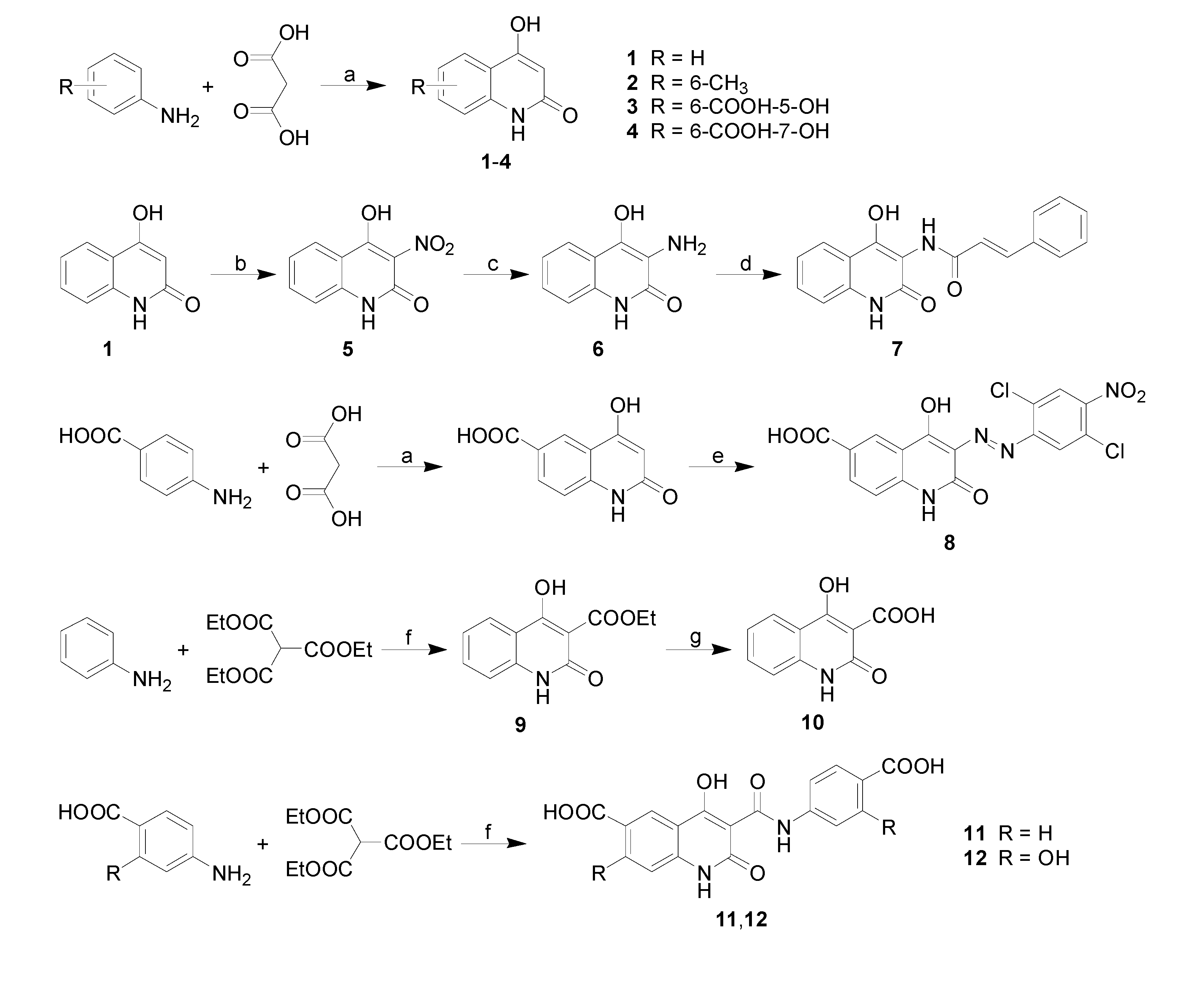
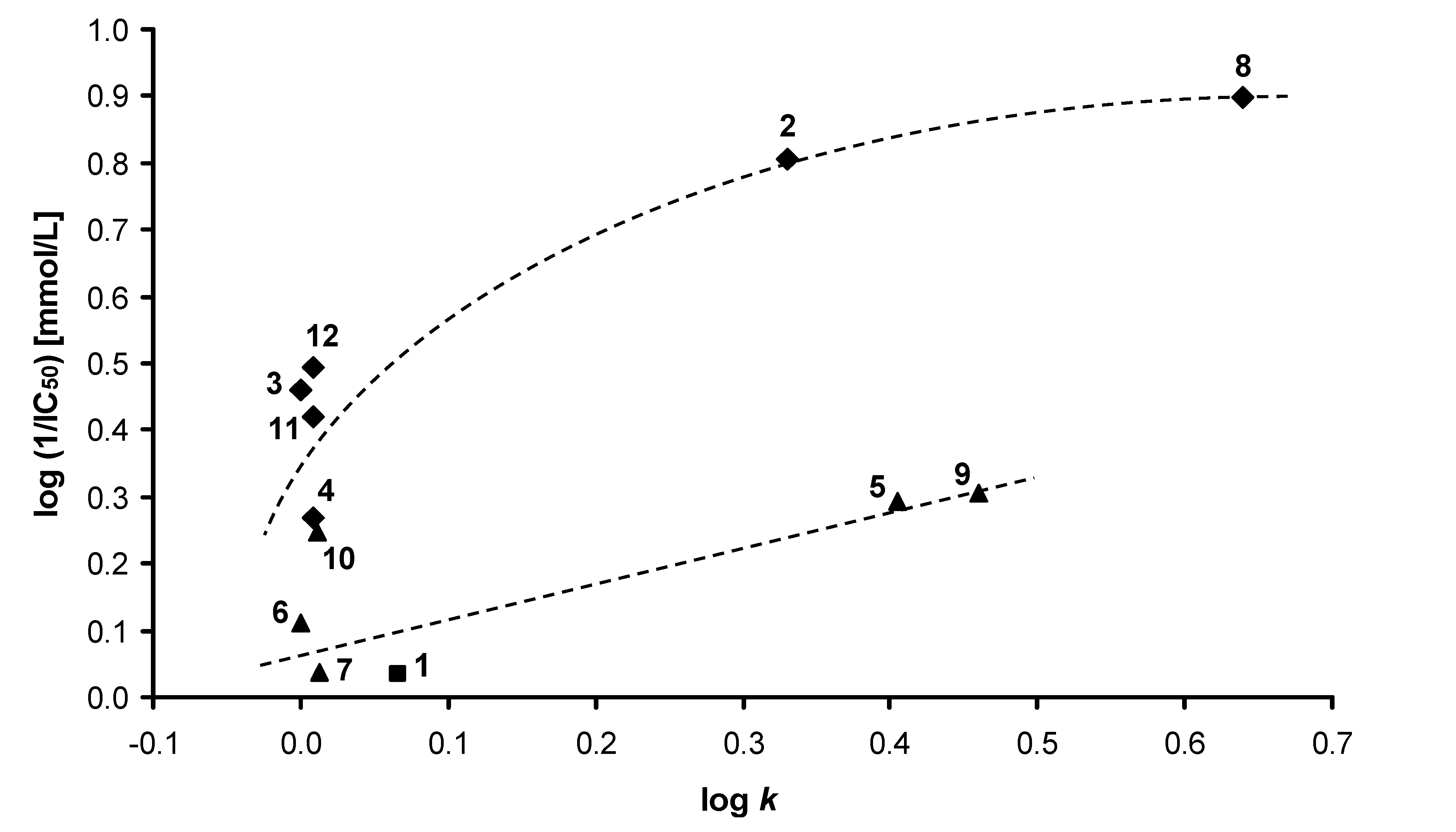
2.2. Lipophilicity
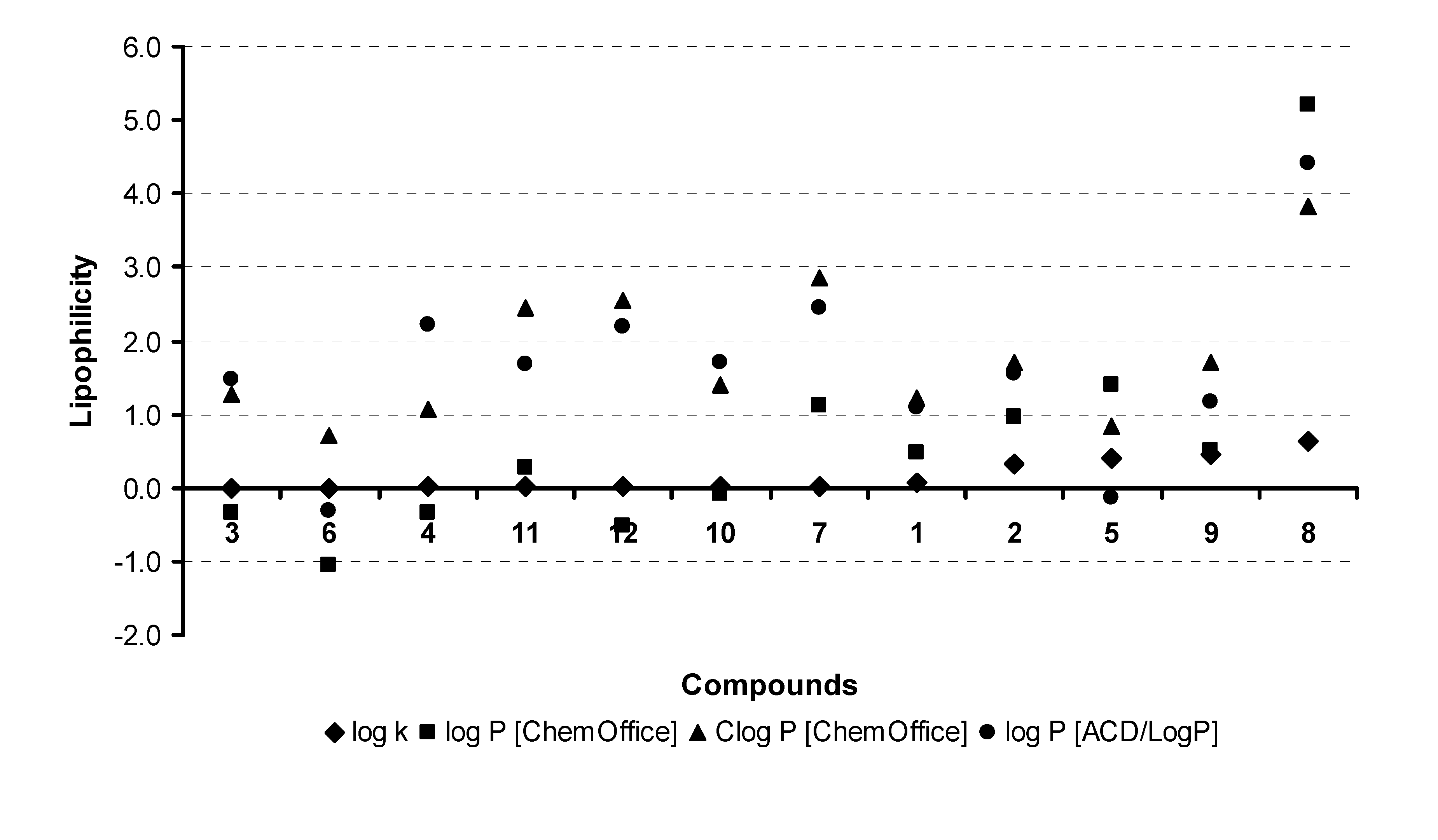

{kind=link}
{kind=link}
{kind=link}
 | |||||
| Comp. | R1 | R2 | log k | log
P/Clog P ChemOffice | log
P ACD/LogP |
|---|---|---|---|---|---|
| 1 | H | H | 0.0664 | 0.49 / 1.216 | 1.10 ± 0.75 |
| 2 | 6-CH3 | H | 0.3307 | 0.97 / 1.715 | 1.56 ± 0.75 |
| 3 | 6-COOH-5-OH | H | 0.0002 | -0.34 / 1.261 | 1.47 ± 0.75 |
| 4 | 6-COOH-7-OH | H | 0.0080 | -0.34 / 1.070 | 2.22 ± 0.75 |
| 5 | H | -NO2 | 0.4052 | 1.39 / 0.836 | -0.14 ± 1.00 |
| 6 | H | -NH2 | 0.0004 | -1.06 / 0.719 | -0.32 ± 1.00 |
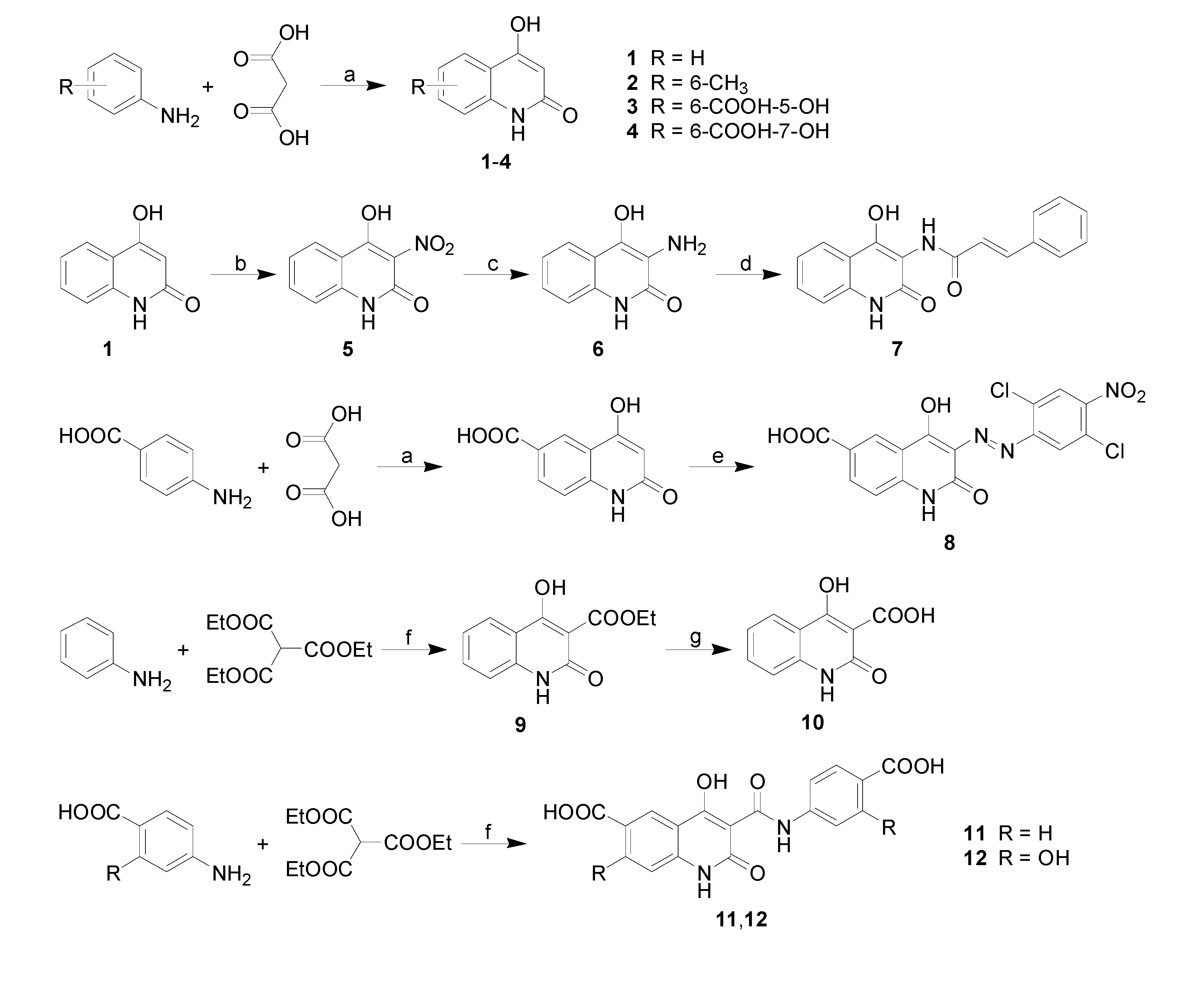
| 7 | H | 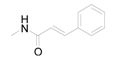 | 0.0128 | 1.11 / 2.848 | 2.45 ± 1.00 |
| 8 | 6-COOH | 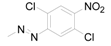 | 0.6394 | 5.22 / 3.840 | 4.41 ± 1.00 |
| 9 | H | -COOC2H5 | 0.4595 | 0.51 / 1.694 | 1.17 ± 0.75 |
| 10 | H | -COOH | 0.0118 | -0.09 / 1.409 | 1.71 ± 0.35 |
| 11 | 6-COOH |  | 0.0081 | 0.27 / 2.445 | 1.67 ± 1.00 |
| 12 | 6-COOH-5-OH |  | 0.0093 | -0.51 / 2.543 | 2.20 ± 1.00 |
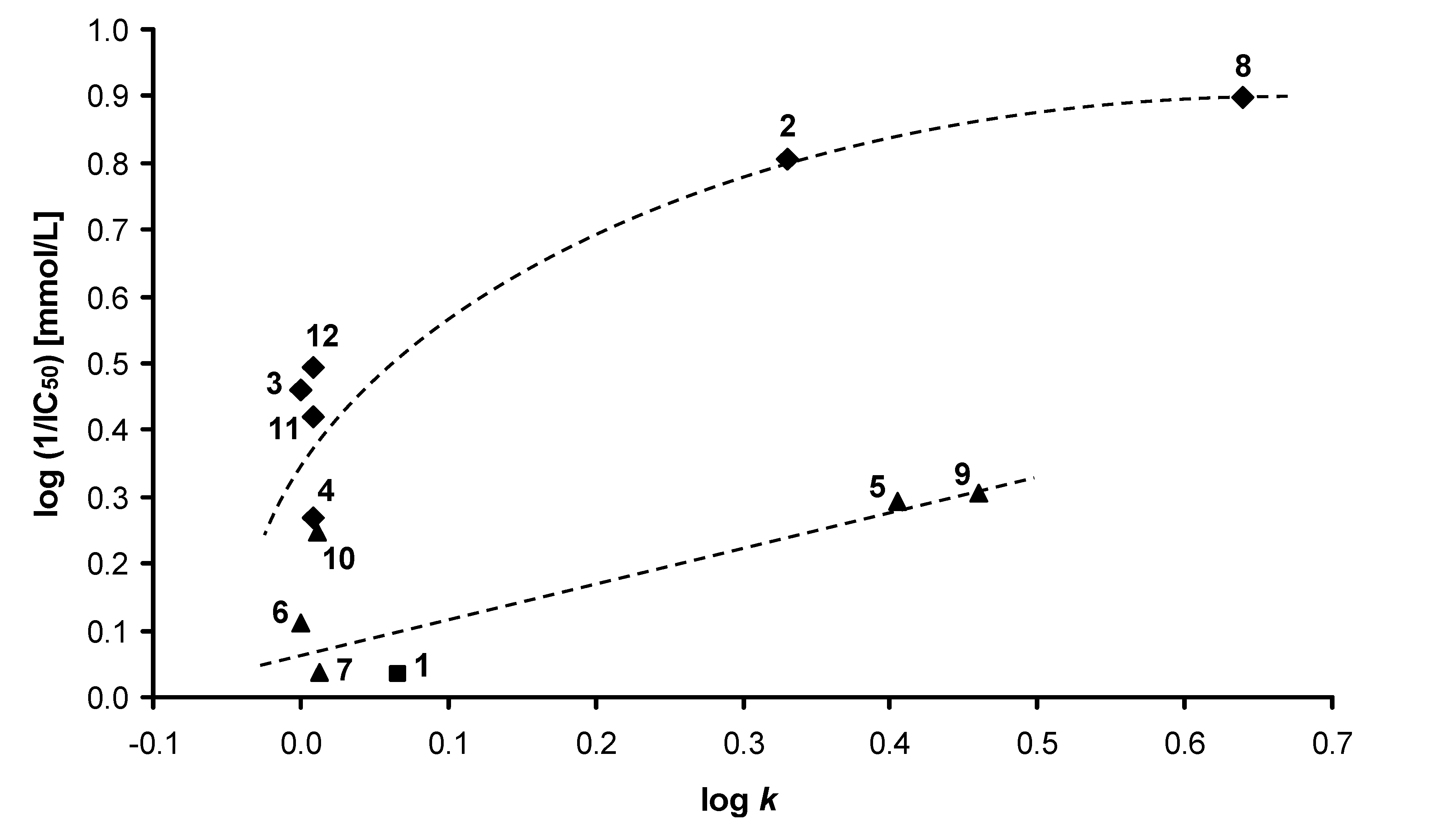
2.3. Oxygen evolution rate inhibition in spinach chloroplasts
| Comp. | MIC/IC80 [µmol/L] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CA | CT | CK | CG | TB | AF | AC | TM | ||
| IC50 [μmol/L] | 24h | 24h | 24h | 24h | 24h | 24h | 24h | 72h | |
| 48h | 48h | 48h | 48h | 48h | 48h | 48h | 120h | ||
| 1 | 925 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | ||
| 2 | 157 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | ||
| 3 | 346 | 125 | 500 | >500 | 250 | 250 | >500 | >500 | >500 |
| 125 | >500 | >500 | 250 | >500 | >500 | >500 | >500 | ||
| 4 | 538 | 15.62 | 500 | >500 | 62.50 | 62.50 | 500 | >500 | >500 |
| 62.50 | >500 | >500 | 250 | >500 | >500 | >500 | >500 | ||
| 5 | 510 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | ||
| 6 | 775 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | ||
| 7 | 916 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 |
| >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | ||
| 8 | 126 | 31.25 | 250 | 250 | 250 | 250 | 125 | 62.50 | 62.50 |
| 125 | >250 | 250 | >250 | >250 | 250 | 250 | 125 | ||
| 9 | 494 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | ||
| 10 | 567 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | ||
| 11 | 380 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | ||
| 12 | 321 | 62.50 | 500 | >500 | 125 | 125 | 500 | 500 | 500 |
| 125 | >500 | >500 | 250 | >500 | >500 | >500 | >500 | ||
| DCMU | 1.9 | - | - | - | - | - | - | - | - |
| FLU | - | 0.06 | 0.12 | 3.91 | 0.98 | 0.24 | >125 | >125 | 1.95 |
| 0.12 | >125 | 15.62 | 3.91 | 0.48 | >125 | >125 | 3.91 | ||
2.4. In vitro antifungal susceptibility testing
3. Conclusions
4. Experimental
4.1. General
4.2. Lipophilicity HPLC determination (capacity factor k / calculated log k)
4.3. Lipophilicity calculations
4.4. Study of inhibition of oxygen evolution rate (OER) in spinach chloroplasts
4.4. In vitro antifungal susceptibility testing
Acknowledgements
References
- Roth, H.J.; Fenner, H. Arzneistoffe, 3rd Ed. ed; Deutscher Apotheker Verlag: Stuttgart, Germany, 2000; pp. 51–114. [Google Scholar]
- Polanski, J.; Niedbala, H.; Musiol, R.; Podeszwa, B.; Tabak, D.; Palka, A.; Mencel, A.; Finster, J.; Mouscadet, J.F.; Le Bret, M. 5-Hydroxy-8-nitro-6-quinaldic Acid as a Novel Molecular Scaffold for HIV-1 Integrase Inhibitors. Lett. Drugs Des. Disc. 2006, 3, 175–178. [Google Scholar] [CrossRef]
- Polanski, J.; Niedbala, H.; Musiol, R.; Podeszwa, B.; Tabak, D.; Palka, A.; Mencel, A.; Mouscadet, J.F.; Le Bret, M. Fragment Based Approach for the Investigation of HIV-1 Integrase Inhibition. Lett. Drugs Des. Disc. 2007, 4, 99–105. [Google Scholar] [CrossRef]
- Vargas, L.Y.; Castelli, M.V.; Kouznetsov, V.V.; Urbina, J.M.; Lopez, S.N.; Sortino, M.; Enriz, R.D.; Ribas, J.C.; Zacchino, S. In vitro Antifungal Activity of New Series of Homoallylamines and Related Compounds with Inhibitory Properties of the Synthesis of Fungal Cell Wall Polymers. Bioorg. Med. Chem. 2003, 11, 1531–1550. [Google Scholar] [CrossRef]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Buchta, V. Quinaldine derivatives preparation and their antifungal activity. ECSOC-8. 2004, p. c005. Available online: http://www.lugo.usc.es/%7Eqoseijas/ECSOC-8/BOCNP/005/index.htm.
- Jampilek, J.; Dolezal, M.; Kunes, J.; Buchta, V.; Kralova, K. Quinaldine Derivatives: Preparation and Biological Activity. Med. Chem. 2005, 1, 591–599. [Google Scholar] [CrossRef]
- Musiol, R.; Jampilek, J.; Buchta, V.; Niedbala, H.; Podeszwa, B.; Palka, A.; Majerz-Maniecka, K.; Oleksyn, B.; Polanski, J. Antifungal Properties of New Series of Quinoline Derivatives. Bioorg. Med. Chem. 2006, 14, 3592–3598. [Google Scholar] [CrossRef]
- Musiol, R.; Jampilek, J.; Kralova, K.; Podeszwa, B.; Finster, J.; Niedbala, H.; Palka, A.; Polanski, J. New quinoline derivatives possessing herbicidal activity. ECSOC-9. 2005, p. c005. Available online: http://www.usc.es/congresos/ecsoc/9/BOCNP/c005/index.htm.
- Musiol, R.; Jampilek, J.; Kralova, K.; Tabak, D.; Podeszwa, B.; Finster, J.; Polanski, J. Substituted amides of quinoline derivatives: preparation and their photosynthesis-inhibiting activity. ECSOC-10. 2006, p. c007. Available online: http://www.usc.es/congresos/ecsoc/10/ECSOC10.htm.
- Musiol, R.; Jampilek, J.; Kralova, K.; Richardson, D.R.; Kalinowski, D.; Podeszwa, B.; Finster, J.; Niedbala, H.; Palka, A.; Polanski, J. Investigating Biological Activity Spectrum for Novel Quinoline Analogues. Bioorg. Med. Chem. 2007, 15, 1280–1288. [Google Scholar] [CrossRef]
- Musiol, R.; Jampilek, J.; Kralova, K.; Tabak, D.; Finster, J.; Podeszwa, B.; Kozik, V.; Dohnal, J.; Polanski, J. Preparation and Herbicidal Activities of Substituted Amides of Quinoline Derivatives. ECSOC-11. 2007, p. a011. Available online: http://www.usc.es/congresos/ecsoc/11/hall_aGOS/a011/index.htm.
- Musiol, R.; Tabak, D.; Niedbala, H.; Podeszwa, B.; Jampilek, J.; Kralova, K.; Dohnal, J.; Finster, J.; Mencel, A.; Polanski, J. Investigating Biological Activity Spectrum for Novel Quinoline Analogues 2: Hydroxyquinolinecarboxamides with Photosynthesis Inhibiting Activity. Bioorg. Med. Chem. 2008, 16, 4490–4499. [Google Scholar] [CrossRef]
- Musiol, R.; Jampilek, J.; Kralova, K.; Finster, J.; Tabak, D.; Niedbala, H.; Csollei, J.; Dohnal, J.; Polanski, J. Ring-substituted 4-hydroxy-1H-quinolin-2-ones: Preparation and Their Photosynthesis-inhibiting Activity. ECSOC-12. 2008, p. c0012. Available online: http://www.usc.es/congresos/ecsoc/12/ECSOC12.htm.
- Podeszwa, B.; Niedbala, H.; Polanski, J.; Musiol, R.; Tabak, D.; Finster, J.; Serafin, K.; Wietrzyk, J.; Boryczka, S.; Mol, W.; Jampilek, J.; Dohnal, J.; Kalinowski, D.; Richardson, D.R. Investigating the Antiproliferative Activity of Quinoline-5,8-dione Analogues on Tumour Cell Lines. Bioorg. Med. Chem. Lett. 2007, 17, 6138–6141. [Google Scholar] [CrossRef]
- Reifler, M.J.; Szalai, V.A.; Peterson, C.N.; Brudvig, G.W. Effects of Tail-like Substituents on the Binding of Competitive Inhibitors to the QB Site of Photosystem II. J. Mol. Recognit. 2001, 14, 157–165. [Google Scholar]
- Moreland, D.E. Research on Biochemistry of Herbicides – an Historical Overview. Z. Naturforsch. C-A J. Biosci. 1993, 48, 121–131. [Google Scholar]
- Zakarya, D.; Larfaoui, E.M.; Boulaamail, A.; Tollabi, M.; Lakhlifi, T. QSARs for a Series of Inhibitory Anilides. Chemosphere 1998, 36, 2809–2818. [Google Scholar] [CrossRef]
- Kralova, K.; Sersen, F.; Kubicova, L.; Waisser, K. Inhibitory Effects of Substituted Benzanilides on Photosynthetic Electron Transport in Spinach Chloroplasts. Chem. Pap. 1999, 53, 328–331. [Google Scholar]
- Dolezal, M.; Miletin, M.; Kunes, J.; Kralova, K. Synthesis and Biological Evaluation of Some Amides of Pyrazine-2-carboxylic acids. Molecules 2002, 7, 363–373. [Google Scholar] [CrossRef]
- Dolezal, M.; Palek, L.; Vinsova, J.; Buchta, V.; Jampilek, J.; Kralova, K. Substituted Pyrazinecarboxamides: Synthesis And Biological Evaluation. Molecules 2006, 11, 242–256. [Google Scholar] [CrossRef]
- Dolezal, M.; Cmedlova, P.; Palek, L.; Vinsova, J.; Kunes, J.; Buchta, V.; Jampilek, J.; Kralova, K. Synthesis and Antimycobacterial Evaluation of Substituted Pyrazinecarboxamides. Eur. J. Med. Chem. 2008, 43, 1105–1113. [Google Scholar]
- Polak, A. The past, Present and Future of Antimycotic Combination Therapy. Mycoses 1999, 42, 355–370. [Google Scholar] [CrossRef]
- Fostel, J.M.; Lartey, P.A. Emerging Novel Antifungal Agents. Drug Discov. Today 2000, 5, 25–32. [Google Scholar] [CrossRef]
- Available online: http://www.doctorfungus.org/ accessed on January 30, 2009.
- Gershon, H; Gershon, M; Clarke, D.D. Synergistic Mixtures of Fungitoxic Monochloro- and Dichloro-8-quinolinols against Five Fungi. Mycopathologia 2004, 158, 131–135. [Google Scholar] [CrossRef]
- Dardari, Z.; Lemrani, M.; Bahloul, A.; Sebban, A.; Hassar, M.; Kitane, S.; Berrada, M.; Boudouma, M. Antileishmanial Activity of a New 8-Hydroxyquinoline Derivative Designed 7-[5′-(3′-phenylisoxazolino)methyl]-8-hydroxyquinoline: preliminary study. Farmaco 2004, 59, 195–199. [Google Scholar] [CrossRef]
- Sheehan, D.J.; Espinel-Ingroff, A.; Steele, M.; Webb, C.D. Antifungal Susceptibility Testing of Yeasts: a Brief Overview. Clin. Infect. Dis. 1993, 17, 494–500. [Google Scholar] [CrossRef]
- Dolezal, M.; Jampilek, J.; Osicka, Z.; Kunes, J.; Buchta, V.; Vichova, P. Substituted 5-aroylpyrazine-2-carboxylic Acid Derivatives: Synthesis and Biological Activity. Farmaco 2003, 58, 1105–1111. [Google Scholar] [CrossRef]
- Collins, J.F.; Donnelly, W.J.; Grundon, M.F.; James, K.J. Biosynthesis of Aromatic Isoprenoids. Part I. The role of 3-Prenylquinolines and of Platydesmine in the Biosnthesis of the Furuquinoline Alkaloid, Dictamnine. J. Chem. Soc., Perkin Trans. 1974, 1, 2177–2181. [Google Scholar]
- Buckle, D.R.; Cantello, B.C.C.; Smith, H.; Spicer, B.A. 4-Hydroxy-3-nitro-2-quinolones and Related Compounds as Inhibitors of Allergic Reactions. J. Med. Chem. 1975, 18, 726–732. [Google Scholar] [CrossRef]
- Ziegler, E.; Wolf, R.; Kappe, T. Synthesen von Heterocyclen, 66. Mitt.: Eine Einfache Synthese des 4-Hydroxycarbostyrils und seiner Derivate. Monatsh. Chem. 1965, 96, 418–422. [Google Scholar] [CrossRef]
- Dolle, V.; Fan, E.; Nguyen, C.H.; Aubertin, A.M.; Kirn, A.; Andreola, M.L.; Jamieson, G.; Tarrago-Litvak, L.; Bisagni, E. A New Series of Pyridinone Derivatives as Potent non-Nucleoside Human Immunodeficiency Virus Type 1 Specific Reverse Transcriptase Inhibitors. J. Med. Chem. 1995, 38, 4679–4686. [Google Scholar] [CrossRef]
- Ukrainets, I.V.; Taran, S.G.; Sidorenko, L.V.; Gorokhova, O.V.; Ogirenko, A.A.; Turov, A.V.; Filimonova, N.I. 4-Hydroxy-2-quinolones. 3-Amino-1-R-2-oxo-4-hydroxyquinolines and Their Acyl Derivatives. Chem. Heterocycl.Compd. 1996, 32, 960–970. [Google Scholar] [CrossRef]
- Ukrainets, I.V.; Gorokhova, O.V.; Sidorenko, L.V. 4-Hydroxyquinol-2-ones. 85. Synthesis of 2-Chloro-4-hydroxyquinoline-3-carboxylic Acid Ethyl Ester. Chem. Heterocycl. Compd. 2005, 41, 1019–1021. [Google Scholar] [CrossRef]
- Detsi, A.; Bardakos, V.; Markopoulos, J.; Igglessi-Markopoulou, O. Reactions of 2-Methyl-3,1-benzoxazin-4-one with Active Methylene Compounds: a New Route to 3-Substituted 4-Hydroxyquinolin-2(1H)-ones. J. Chem. Soc., Perkin Trans. 1 1996, 24, 2909–2913. [Google Scholar]
- Masarovicova, E.; Kralova, K. Approaches to Measuring Plant Photosynthesis Activity. In Handbook of Photosynthesis, 2nd Ed.; Pessarakli, M., Ed.; Taylor & Francis Group: Boca Raton, London-New York-Singapore, 2005; pp. 617–656. [Google Scholar]
- Kralova, K.; Sersen, F.; Sidoova, E. Photosynthesis Inhibition Produced by 2-Alkylthio-6-R-benzothiazoles. Chem. Pap. 1992, 46, 348–350. [Google Scholar]
- Fedke, C. Biochemistry and Physiology of Herbicide Action.; Springer Verlag: Berlin-Heidelberg-New York, USA , 1982. [Google Scholar]
- National Committee for Clinical Laboratory standards. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeast: Approved Standard, NCCLS document M27-A; NCCLS: Villanova, PA, USA, 1997. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jampilek, J.; Musiol, R.; Pesko, M.; Kralova, K.; Vejsova, M.; Carroll, J.; Coffey, A.; Finster, J.; Tabak, D.; Niedbala, H.; et al. Ring-substituted 4-Hydroxy-1H-quinolin-2-ones: Preparation and Biological Activity. Molecules 2009, 14, 1145-1159. https://doi.org/10.3390/molecules14031145
Jampilek J, Musiol R, Pesko M, Kralova K, Vejsova M, Carroll J, Coffey A, Finster J, Tabak D, Niedbala H, et al. Ring-substituted 4-Hydroxy-1H-quinolin-2-ones: Preparation and Biological Activity. Molecules. 2009; 14(3):1145-1159. https://doi.org/10.3390/molecules14031145
Chicago/Turabian StyleJampilek, Josef, Robert Musiol, Matus Pesko, Katarina Kralova, Marcela Vejsova, James Carroll, Aidan Coffey, Jacek Finster, Dominik Tabak, Halina Niedbala, and et al. 2009. "Ring-substituted 4-Hydroxy-1H-quinolin-2-ones: Preparation and Biological Activity" Molecules 14, no. 3: 1145-1159. https://doi.org/10.3390/molecules14031145
APA StyleJampilek, J., Musiol, R., Pesko, M., Kralova, K., Vejsova, M., Carroll, J., Coffey, A., Finster, J., Tabak, D., Niedbala, H., Kozik, V., Polanski, J., Csollei, J., & Dohnal, J. (2009). Ring-substituted 4-Hydroxy-1H-quinolin-2-ones: Preparation and Biological Activity. Molecules, 14(3), 1145-1159. https://doi.org/10.3390/molecules14031145