General
Melting points were determined on a Stuart Scientific SMP 2 melting point apparatus and are uncorrected. Infrared spectra were recorded on CH2Cl2-films with a Perkin Elmer Spectrum GX FT-IR spectrophotometer. Ultraviolet spectra were recorded on methanol solutions with a Hitachi U-3300 spectrophotometer. 1H- and 13C-NMR spectra were recorded for deuterochloroform solutions at 300 MHz for 1H and 75 MHz for 13C with a Bruker AVANCE 300 spectrometer. Tetramethylsilane was used as the internal standard. High Resolution Mass spectra were recorded with a Bruker Daltonics MicrOTOF mass spectrometer.
N-(3,4-Dimethoxyphenethyl)-2-(2-bromophenyl)acetamide (
8a). A mixture of 2-bromophenylacetic acid (15.0 g, 0.07 mol) and thionyl chloride (20.8 g) in benzene (50 mL) was refluxed for 1 h. Removal of the solvent under vacuum gave 2-bromophenylacetyl chloride (
7) which was dissolved in ethanol-free chloroform (50 mL) and added to a mixture of 3,4-dimethoxyphenethylamine (
6a, 12.7 g, 0.07 mol) in chloroform (100 mL) and 10% sodium hydrogen carbonate (100 mL). The mixture was then stirred for 3 h and the chloroform layer was washed with water (2 × 100 mL), 10% hydrochloric acid (3 × 50 mL), water (100 mL), then dried over anhydrous sodium sulfate. Removal of the solvent under vacuum gave a residue which was recrystallized from ethanol to give amide
8a as a pale yellow solid (22.0 g, 88.1%); m.p. 131-132 °C (Lit. [
15] m.p. 127-129 °C);
1H-NMR: δ 7.54 (1H, d,
J = 7.8 Hz, Ar-H); 7.30-7.22 (2H, m, Ar-H); 7.18-7.09 (1H, m, Ar-H); 6.72 (1H, d,
J = 8.1 Hz, Ar-H); 6.63 (1H, d,
J = 1.9 Hz, Ar-H); 6.60 (1H, dd,
J = 8.1, 1.9 Hz, Ar-H); 5.58 (1H, s, NH); 3.85 (3H, s, OCH
3); 3.82 (3H, s, OCH
3); 3.66 (2H, s, CH
2); 3.47 (2H, apparent q,
J = 6.8 Hz, CH
2); 2.71 (2H, apparent t,
J = 7.0 Hz, CH
2);
13C-NMR: δ 169.48(C), 148.98(C), 147.60(C), 134.79(C), 133.03(CH), 131.63(CH), 131.10(C), 129.04(CH), 127.92(CH), 124.94(C), 120.58(CH), 111.79(CH), 111.32(CH), 55.91(OCH
3), 55.82 (OCH
3), 43.99(CH
2), 40.81(CH
2), 34.99(CH
2).
N-(2,3,4-Trimethoxyphenethyl)-2-(2-bromophenyl)acetamide (8b). In a similar manner, 8b was obtained in 78.8% yield as a pale yellow solid from ethanol; m.p. 92-93 °C; 1H-NMR: δ 7.57 (1H, d, J = 8.3 Hz, Ar-H); 7.33-7.25 (2H, m, Ar-H); 7.19-7.12 (1H, m, Ar-H); 6.72 (1H, d, J = 8.5 Hz, Ar-H); 6.54 (1H, d, J = 8.5 Hz, Ar-H); 5.83 (1H, br s, NH); 3.88 (3H, s, OCH3); 3.83 (3H, s, OCH3); 3.79 (3H, s, OCH3); 3.67 (2H, s, CH2); 3.43 (2H, apparent q, J = 6.4 Hz, CH2); 2.70 (2H, apparent t, J = 6.7 Hz, CH2); 13C-NMR: δ 169.58(C), 152.59(C), 151.74(C), 142.19(C), 134.91(C), 133.05(CH), 131.78(CH), 128.97(CH), 127.89(CH), 125.10(C), 124.69(C), 124.46(CH), 107.42(CH), 60.92(OCH3), 60.75(OCH3), 56.01(OCH3), 44.05 (CH2), 40.91(CH2), 29.47(CH2).
N-(3,4-Methylenedioxyphenethyl)-2-(2-bromophenyl)acetamide (
8c). In a similar manner,
8c was obtained in 82.5% yield as a pale yellow solid from ethanol; m.p. 124-126 °C (Lit. [
16] m.p. 128-130 °C);
1H-NMR: δ 7.56 (1H, d,
J = 7.8 Hz, Ar-H); 7.33-7.25 (2H, m, Ar-H); 7.19-7.11 (1H, m, Ar-H); 6.65 (1H, d,
J = 7.9 Hz, Ar-H); 6.55 (1H, d,
J = 1.6 Hz, Ar-H); 6.48 (1H, dd,
J = 7.9, 1.6 Hz, Ar-H); 5.91 (2H, s, OCH
2O); 5.48 (1H, br s, NH); 3.66 (2H, s, CH
2); 3.42 (2H, apparent q,
J = 6.7 Hz, CH
2); 2.65 (2H, apparent t,
J = 6.7 Hz, CH
2);
13C-NMR: δ 169.44(C), 147.72(C), 146.11(C), 134.80(C), 133.13(CH), 132.35(C), 131.67(CH), 129.09(CH), 127.97(CH), 124.98(C), 121.57(CH), 109.04(CH), 108.30(CH), 100.85(CH
2), 44.04(CH
2), 40.87(CH
2), 35.15(CH
2).
1-(2-Bromobenzyl)-3,4-dihydro-6,7-dimethoxyisoquinoline (
9a). A solution of
8a (5.5 g, 14.6 mmol) and phosphorus oxychloride (17.0 g) in benzene (60 mL) was refluxed for 3 h. The excess reagent and solvent were removed under vacuum. The residue was shaken with chloroform (100 mL) and dilute ammonium hydroxide (100 mL). The chloroform layer was washed with water (100 mL), then dried over anhydrous sodium carbonate. Removal of the solvent under vacuum gave dihydroisoquinoline
9a as a pale yellow solid (3.9 g, 75.8%) from ethyl acetate-hexane; m.p. 95-96 °C (Lit. [
15] m.p. 93-95 °C). It was found to be unstable and was immediately used in the next step without further purification.
1H-NMR: δ 7.56 (1H, dd,
J = 7.9, 1.3 Hz, Ar-H); 7.27 (1H, dd,
J = 7.6, 1.7 Hz, Ar-H); 7.18 (1H, dt,
J = 7.6, 1.3 Hz, Ar-H); 7.05 (1H, dt,
J = 7.6, 1.7 Hz, Ar-H); 6.91 (1H, s, Ar-H); 6.67 (1H, s, Ar-H); 4.20 (2H, s, Ar-CH
2); 3.88 (3H, s, OCH
3); 3.79 (3H, s, OCH
3); 3.73 (2H, t,
J = 7.6 Hz, CH
2); 2.67 (2H, t,
J = 7.6 Hz, CH
2);
13C-NMR δ: 165.08(C), 150.76(C), 147.43(C), 137.66(C), 132.78(CH), 131.59(C), 130.21 (CH), 128.18(CH), 127.62(CH), 124.54(C), 121.41(C), 110.25(CH), 109.15(CH), 56.16(OCH
3), 55.92(OCH
3), 47.29(CH
2), 42.53(CH
2), 25.73(CH
2).
1-(2-Bromobenzyl)-3,4-dihydro-5,6,7-trimethoxyisoquinoline (9b). In a similar manner, 9b was obtained in almost quantitative yield as a yellow viscous oil. 1H-NMR: δ 7.55 (1H, dd, J = 7.9, 1.2 Hz, Ar-H); 7.30-7.26 (1H, m, Ar-H); 7.20-7.15 (1H, m, Ar-H); 7.09-7.01 (1H, m, Ar-H); 6.78 (1H, s, H−8); 4.19 (2H, s, Ar−CH2); 3.88 (3H, s, OCH3); 3.83 (3H, s, OCH3); 3.78 (3H, s, OCH3); 3.71 (2H, t, J = 7.6 Hz, CH2); 2.67 (2H, t, J = 7.6 Hz, CH2); 13C-NMR: δ 164.68(C), 151.69(C), 149.88(C), 144.17 (C), 137.63(C), 132.77(CH), 130.23(CH), 128.19(CH), 127.61(CH), 124.52(C), 124.46(C), 124.09(C), 105.63(CH), 60.88(OCH3), 60.83(OCH3), 56.19(OCH3), 47.09 (CH2), 42.58(CH2), 18.99(CH2).
1-(2-Bromobenzyl)-3,4-dihydro-6,7-methylenedioxyisoquinoline (
9c). In a similar manner,
9c was obtained in 42.1% yield from ethanol as a pale yellow solid; m.p. 122-123 °C (Lit. [
16] m.p. 121-123 °C);
1H-NMR: δ 7.52 (1H, dd,
J = 7.8, 1.0 Hz, Ar-H); 7.22-7.12 (2H, m, Ar-H); 7.06- 6.98 (1H, m, Ar-H); 6.89 (1H, s, Ar-H); 6.61 (1H, s, Ar-H); 5.86 (2H, s, OCH
2O); 4.09 (2H, s, Ar-CH
2); 3.65 (2H, t,
J = 7.6 Hz, CH
2); 2.60 (2H, t,
J = 7.6 Hz, CH
2);
13C-NMR: δ 164.65(C), 149.08(C), 146.44(C), 137.55(C), 133.38(C), 132.79 (CH), 130.30(CH), 128.14(CH), 127.50(CH), 124.79(C), 122.77(C), 107.98(CH), 106.02(CH), 101.31(CH
2), 47.09(CH
2), 42.55(CH
2), 26.33(CH
2).
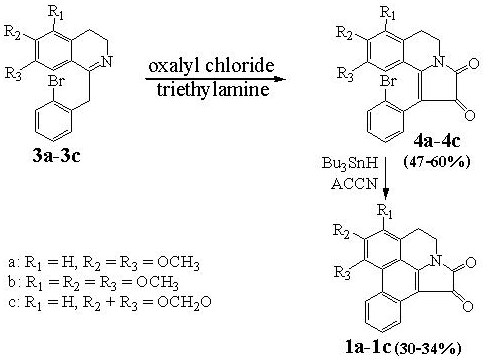
1-(2-Bromophenyl)-5,6-dihydro-8,9-dimethoxypyrrolo[2,1-a]isoquinoline-2,3-dione (
10a). Oxalyl chloride (0.2 mL) was added dropwise to a stirred solution of
9a (359 mg, 1 mmol), triethylamine (0.3 mL) in chloroform (10 mL) at room temperature. Stirring was continued for 3 h. Chloroform (20 mL) was added and the chloroform layer was washed with 5% hydrochloric acid (4 × 50 mL), water (50 mL), then dried over anhydrous sodium sulfate. Removal of the solvent under vacuum gave a residue which was recrystallized from ethanol to give 10a as red prisms (247.8 mg, 60.0%); m.p. 195-196 °C. (lit. [
6] m.p. 176-178 °C); UV λ
max (MeOH) nm (log ε): 203 (4.36), 226sh (4.12), 262 (3.76), 322 (3.65). IR (CH
2Cl
2-film) ν
max cm
-1: 2937, 2843, 1744, 1699, 1594, 1575, 1515, 1472, 1428, 1398, 1337, 1312, 1291, 1270, 1225, 1187, 1101, 1034, 987, 865, 798, 735, 683;
1H-NMR: δ 7.70 (1H, dd,
J = 8.0, 0.9 Hz, Ar-H); 7.44-7.37 (1H, m, Ar-H); 7.34-7.29 (1H, m, Ar-H); 7.29-7.22 (1H, m, Ar-H); 6.77 (1H, s, Ar-H); 6.66 (1H, s, Ar-H); 3.95 (3H, s, OCH
3); 3.89 (2H, t,
J = 6.3 Hz, CH
2); 3.29 (3H, s, OCH
3); 3.10 (2H, t,
J = 6.3 Hz, CH
2);
13C-NMR: δ 181.59(C), 158.50(C), 158.35(C), 153.80(C), 148.14(C), 133.17(CH), 133.09(C), 132.83(CH), 132.41(C), 129.96(CH), 128.06(CH), 125.82(C), 116.68(C), 111.29(CH), 107.80(C), 56.27(OCH
3), 55.17(OCH
3), 36.37(CH
2), 28.37(CH
2). HRMS (ESI-TOF) calcd for C
20H
16BrNO
4 ([M+H
+]) = 414.0335, Found 414.0438.
1-(2-Bromophenyl)-5,6-dihydro-7,8,9-trimethoxypyrrolo[2,1-a]isoquinoline-2,3-dione (10b). In a similar manner, 10b was obtained as a deep red solid in 68.2% yield after chromatography over alumina using dichloromethane as eluent; m.p. 69-70 °C; UV (MeOH) λ max nm (log ε): 203 (4.66), 226sh (4.38), 258 (3.95), 332 (3.93); IR (CH2Cl2-film) νmax cm-1: 2939, 2837, 1746, 1702, 1592, 1576, 1467, 1425, 1397, 1342, 1298, 1248, 1182, 1109, 1024, 986, 939, 914, 845, 752; 1H-NMR: δ 7.71 (1H, dd, J = 8.0, 0.9 Hz, Ar-H); 7.44-7.35 (1H, m, Ar-H); 7.32-7.23 (2H, m, Ar-H); 6.53 (1H, s, Ar-H); 3.94 (3H, s, OCH3); 3.88 (3H, s, OCH3); 3.93-3.73 (2H, m, CH2); 3.27 (3H, s, OCH3); 3.20-3.00 (2H, m, CH2); 13C-NMR: δ 182.05(C), 158.07(C), 152.24(C), 150.56(C), 147.02(C), 133.21(CH), 132.70(CH), 132.25(C), 130.03(CH), 128.08(CH), 125.77(C), 125.48(C), 119.38(C), 108.64(C), 108.53(CH), 105.68(C), 61.12(OCH3), 61.08(OCH3), 55.21(OCH3), 36.19(CH2), 21.67(CH2). HRMS (ESI-TOF) calcd for C21H18BrNO5 ([M+H+]) = 444.0441, Found 444.0519.
1-(2-Bromophenyl)-5,6-dihydro-8,9-methylenedioxypyrrolo[2,1-a]isoquinoline-2,3-dione (10c). In a similar manner, 10c was obtained in 47.7% yield from ethanol as a deep red prisms; m.p. 226-227 °C; UV (MeOH) λ max nm (log ε): 203 (4.64), 236sh (4.18), 261sh (3.88), 284 (3.77), 320 (3.67), 388 (3.64); IR (CH2Cl2-film) νmax cm-1: 3056, 2906, 1744, 1698, 1608, 1568, 1505, 1467, 1403, 1378, 1338, 1316, 1286, 1249, 1181, 1036, 938, 868, 748, 736; 1H-NMR: δ 7.71-7.66 (1H, m, Ar-H); 7.42-7.36 (1H, m, Ar-H); 7.31-7.23 (2H, m, Ar-H); 6.79 (1H, s, Ar-H); 6.56 (1H, s, Ar-H); 6.00 (2H, AB q, J = 1.1 Hz, OCH2O); 3.94-3.77 (2H, m, CH2); 3.07 (2H, t, J = 6.3 Hz, CH2); 13C-NMR: δ 181.94(C), 158.10(C), 157.94(C), 152.47(C), 147.37(C), 135.17(C), 133.43(CH), 132.43(CH), 131.68(C), 130.13(CH), 128.14(CH), 125.35(C), 118.23(C), 109.28(CH), 108.50(CH), 108.23(C), 102.25(CH2), 36.20(CH2), 29.24(CH2). HRMS (ESI-TOF) calcd for C19H12BrNO4 ([M+H+]) = 398.0022, Found 397.9895.
Telisatin A (
1). A solution of 1,1ʹ-azobis(cyclohexanecarbonitrile) (245.0 mg, 1.0 mmol) and tributyltin hydride (1.2 g, 4.0 mmol) in toluene (20 mL) was added dropwise in four equal portions over 3 h to a refluxing solution of
10a (413.0 mg, 1.0 mmol) in toluene (20 mL) and the resulting mixture was then refluxed for another 8 h. The solvent was then removed under vacuum and the residue was dissolved in acetonitrile (40 mL) and washed with hexane (2 × 30 mL), then dried over anhydrous sodium sulfate. Removal of the solvent gave a brown viscous oil (0.4 g) which was recrystallized with ethanol to give telisatin A (
1) as red prisms (109.9 mg, 33.0%); m.p. 234-235 °C (Lit. [
1] m.p. 238-239 °C); UV (MeOH) λ
max nm (log ε): 207 (4.03), 257 (4.26), 284sh (3.60), 322 (3.70), 336 (3.80), 352sh (3.56); IR (CH
2Cl
2-film) ν
max cm
-1: 2925, 1748, 1701, 1605, 1584, 1531, 1462, 1423, 1386, 1306, 1261, 1195, 1149, 1131, 1112, 1037, 969, 924, 802, 759.
1H-NMR δ: 9.41 (1H, br d,
J = 8.5 Hz, H−11); 8.63 (1H, dd,
J = 8.0, 1.5 Hz, H−8); 7.67-7.60 (1H, m, H−9); 7.54-7.46 (1H, m
, H−10); 7.17 (1H, s, H−3); 4.10 (3H, s, OCH
3); 3.97 (2H, t,
J = 6.5 Hz, CH
2); 3.95 (3H, s, OCH
3); 3.35 (2H, t,
J = 6.5 Hz, CH
2);
13C-NMR: δ 179.98(C), 160.34(C), 157.15(C), 153.36(C), 146.65(C), 130.75(C), 129.35(C), 129.21(CH), 128.33(CH), 127.56(C), 125.87(C), 125.62(CH), 123.76(CH), 112.29(CH), 112.18(C), 103.17(C), 59.99(OCH
3), 56.62(OCH
3), 36.53(CH
2), 27.68 (CH
2). HRMS (ESI-TOF) calcd for C
20H
15NO
4 ([M+H
+]) = 334.1074, Found 334.1125.
Telisatin B (
2). In a similar manner, telisatin B (
2) was obtained as deep red prisms (30.0%) from ethanol; m.p. 218-219 °C (Lit.[
1] m.p. 221-222 °C); UV (MeOH) λ
max nm (log ε): 203 (4.33), 223sh (4.26), 257 (4.59), 318sh (3.99), 329 (4.07); IR (CH
2Cl
2-film) ν
max cm
-1: 2942, 2864, 1749, 1716, 1702, 1619, 1607, 1579, 1527, 1515, 1452, 1406, 1389, 1323, 1146, 1125, 1071, 1033, 973, 814, 757;
1H-NMR: δ 9.37 (1H, br d,
J = 8.5 Hz, H−11), 8.60 (1H, dd,
J = 8.0, 1.3 Hz, H−8), 7.64-7.56 (1H, m, H−9), 7.55-7.45 (1H, m, H−10), 4.14 (3H, s, OCH
3), 4.03 (3H, s, OCH
3), 4.01 (3H, s, OCH
3), 3.91 (2H, t,
J = 6.5 Hz, CH
2), 3.33 (2H, t,
J = 6.5 Hz, CH
2);
13C-NMR: δ 180.69(C), 159.86(C), 152.58(C), 152.16(C), 152.03(C), 150.16(C), 128.71(CH), 127.53(CH), 126.63(C), 126.38(C), 126.11(C), 125.78(CH), 123.66(CH), 121.18(C), 114.14(C), 104.17(C), 61.47(OCH
3), 61.30(OCH
3), 60.48 (OCH
3), 36.14(CH
2), 21.21(CH
2); HRMS (ESI-TOF) calcd for C
21H
17NO
5 ([M+H
+]) = 364.1179, Found 364.1231.
Lettowianthine (
3). In a similar manner, lettowianthine (
3) was obtained as red prisms (34.0%); m.p. 294-295 °C (dec.) (Lit. [
2] m.p. 314-317 °C (dec.); Lit.[
4] m.p. 265-267 °C); UV (MeOH) λ
max nm (log ε): 203 (4.37), 212sh (4.29), 247sh (4.07), 257sh (4.04), 287 (3.64), 335 (3.51), 353 (3.28). IR (CH
2Cl
2-film) ν
max cm
-1: 2923, 2093, 1737, 1695, 1622, 1610, 1581, 1530, 1506, 1450, 1417, 1301, 1253, 1222, 1176, 1151, 1122, 1050, 927, 867, 749;
1H-NMR: δ 8.80 (1H, br d,
J = 8.6 Hz, H−11), 8.55 (1H, br d,
J = 7.6 Hz, H−8), 7.61 (1H, t,
J = 6.9 Hz, H−9), 7.48 (1H, t,
J = 7.2 Hz, H−10), 7.09 (1H, s, H−3), 6.35 (2H, s, OCH
2O), 3.93 (2H, t,
J = 6.2 Hz, CH
2), 3.29 (2H, t,
J = 6.2 Hz, CH
2);
13C-NMR δ: 179.92(C), 160.33(C), 153.68(C), 151.55(C), 143.08(C), 129.61(C), 129.33(CH), 127.62 (CH), 126.83(C), 125.35(CH), 124.45(C), 123.60(CH), 119.90(C), 112.46(C), 109.23(CH), 103.10 (C), 102.35(CH
2), 36.66(CH
2), 27.51(CH
2); HRMS (ESI-TOF) calcd for C
19H
11NO
4 ([M+H
+]) = 318.0761, Found 318.0666.

{kind=link}
{kind=link}
{kind=link}

