New Macrocyclic Amines Showing Activity as HIV Entry Inhibitors Against Wild Type and Multi-Drug Resistant Viruses

 ,
,  and
and
Abstract
:1. Introduction

2. Results and Discussion
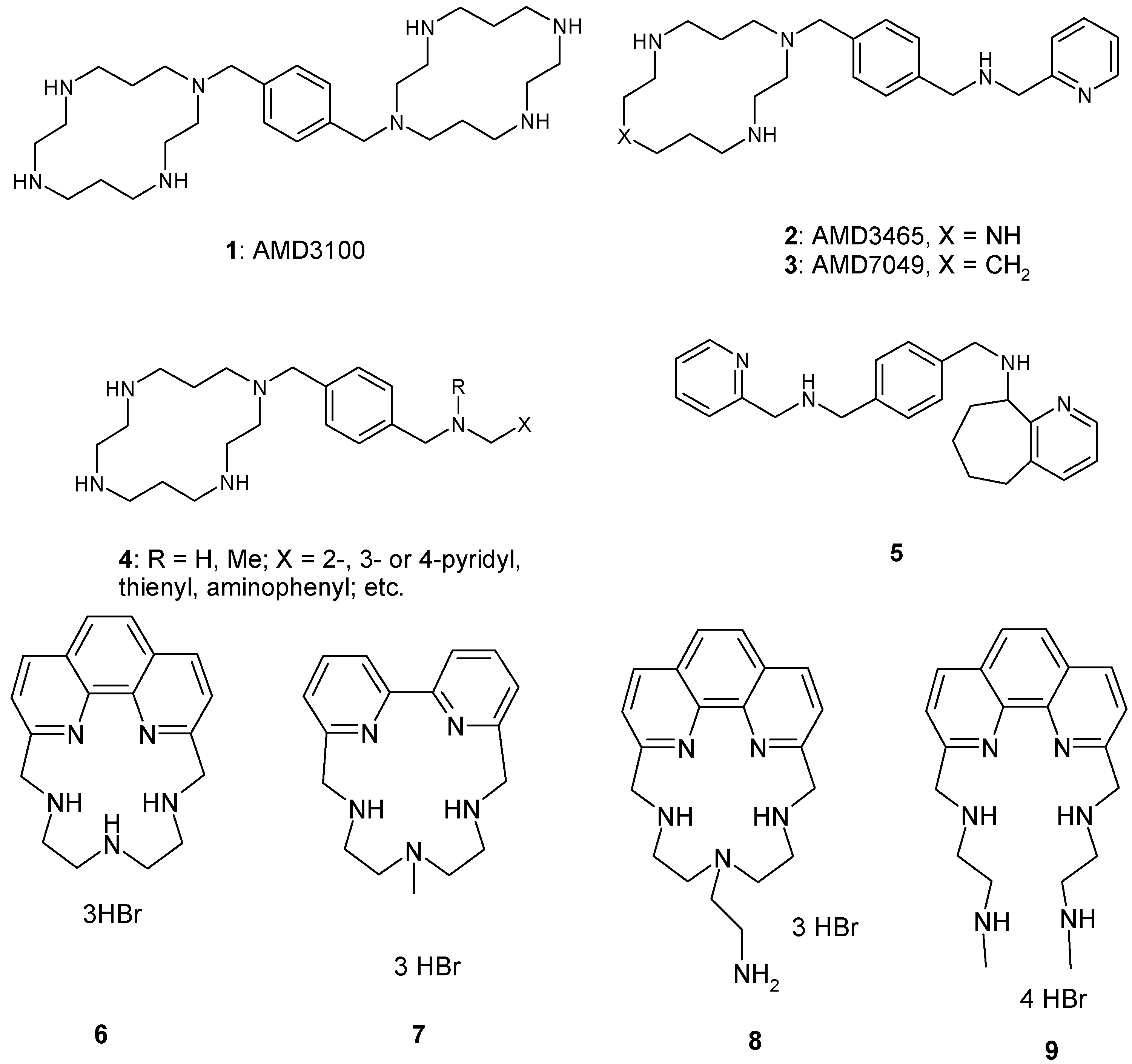
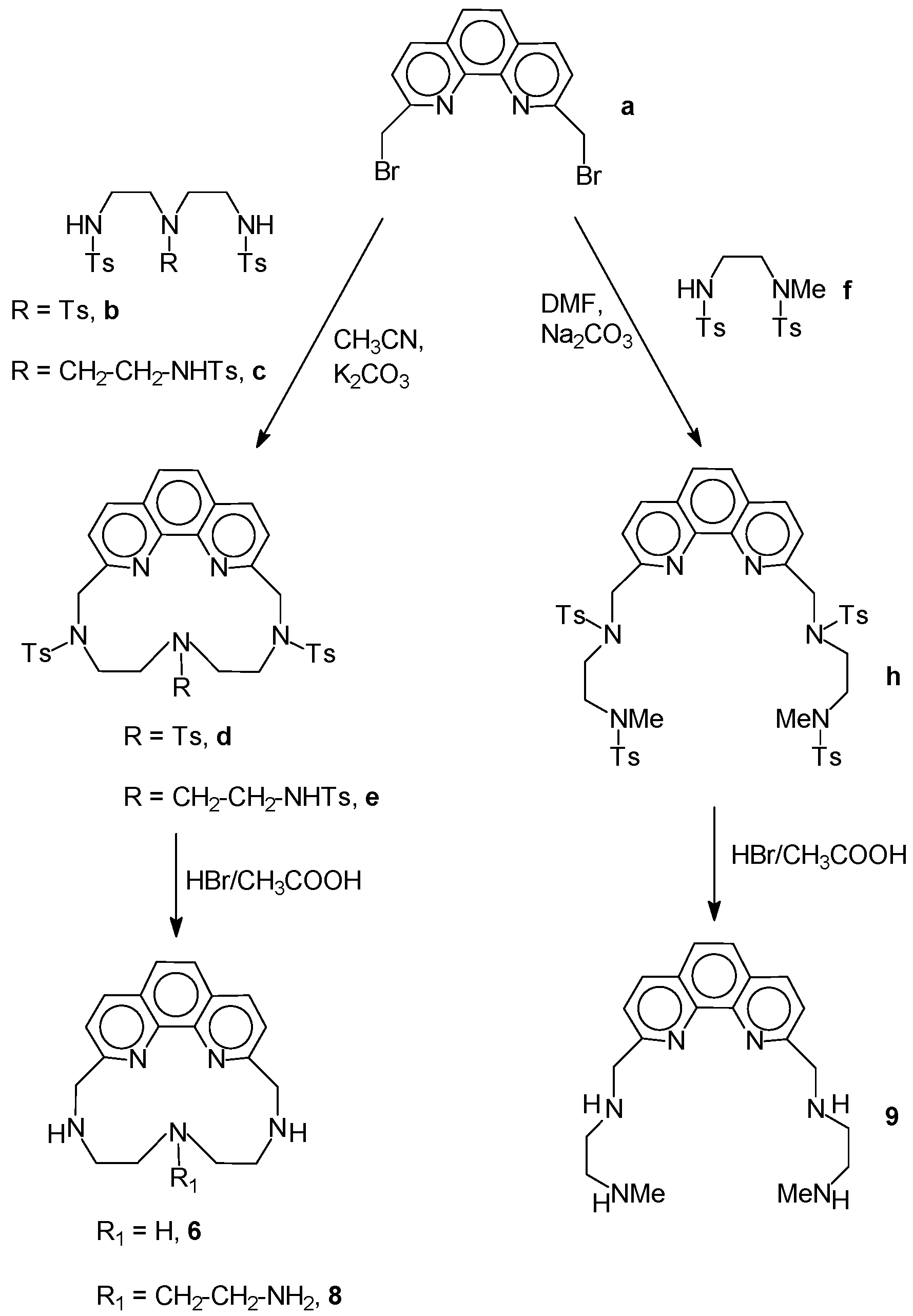
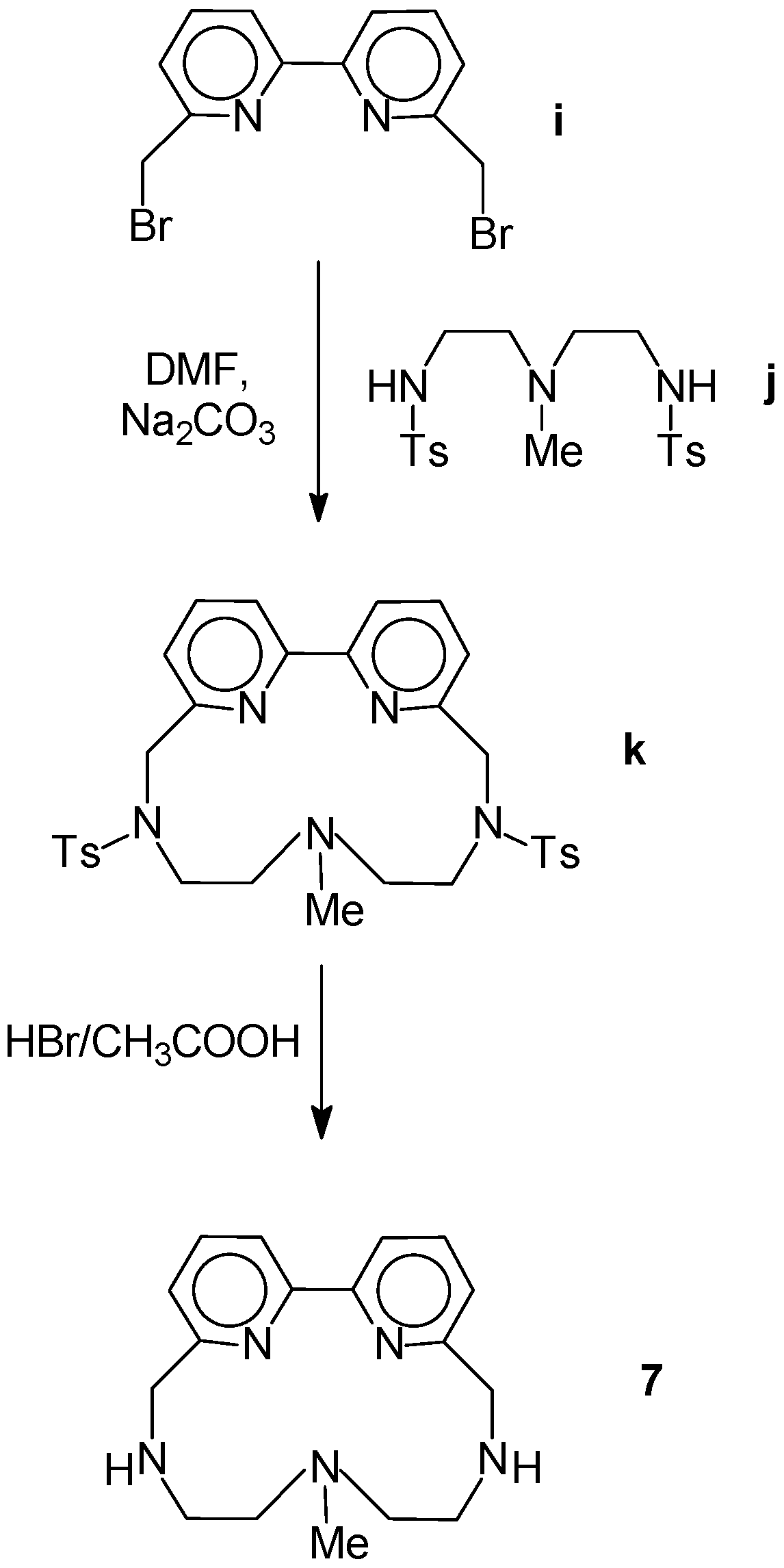
2.1. Chemistry


2.2. Definition of co-receptor usage
2.3. Antiviral assay
2.3.1. In vitro antiviral activity on a wild type HIV strain

{kind=link}
{kind=link}
{kind=link}
| Compound | HIV-1 (14aPre)a | |
|---|---|---|
| IC50(µM)b | SEM (µM)c | |
| AMD3100 | 0.829 | 1.304 |
| Compound #6 | 2.595 | 0.615 |
| Compound #7 | 2.436 | 1.304 |
| Compound #8 | 3.511 | 1.144 |
| Compound #9 | 3.084 | 1.001 |
2.3.2. In vitro antiviral activity on dual tropic isolates
| Patienta | IC50 values (week 0)b | IC50 values (week 24 - 48)c |
|---|---|---|
| TO1 | 2.43 - 3.51 µM | 2.62 - 3.33 µM |
| T03 | 2.36 - 3.22 µM | 2.72 - 3.34 µM |
| RCP | 2.63 - 3.55 µM | 2.72 - 3.34 µM |
3. Experimental
3.1. Anti-HIV activity assays
3.1.1. Viruses
3.1.2. Co-receptor usage
3.1.3. Compounds formulation
3.1.4. Susceptibility assays
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- UNAIDS/WHO. AIDS Epidemic Update: December 2008. Available online: http://www.unaids.org.
- Graves, M.C.; Lim, J.J.; Heimer, E.P.; Kramer, R.A. An 11-kDa form of human immunodeficiency virus protease expressed in Escherichia coli is sufficient for enzymatic activity. Proc. Natl. Acad. Sci. USA 1988, 85, 2449–2453. [Google Scholar]
- Drake, S.M. NNRTIs-a new class of drugs for HIV. J. Antimicr. Chemother. 2000, 45, 417–420. [Google Scholar] [CrossRef]
- De Clercq, E. New approaches toward anti-HIV chemotherapy. J. Med. Chem. 2005, 48, 1297–1313. [Google Scholar] [CrossRef]
- Barbaro, G.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Highly active antiretroviral therapy: current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome. Curr. Pharm. Des. 2005, 11, 1805–1843. [Google Scholar] [CrossRef]
- Grabar, S.; Pradier, C.; Le Corfec, E.; Lancar, R.; Allavena, C.; Bentata, M.; Berlureau, P.; Dupont, C.; Fabbro-Peray, P.; Poizot-Martin, I.; Costagliola, D. Factors associated with clinical and virological failure in patients receiving a triple therapy including a protease inhibitor. AIDS 2000, 14, 141–149. [Google Scholar] [CrossRef]
- Citterio, P.; Rusconi, S. Novel inhibitors of the early steps of the HIV-1 life cycle. Exp. Opin. Invest. Drugs 2007, 16, 11–23. [Google Scholar] [CrossRef]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef]
- Cocchi, F.; De Vico, A.L.; Garzino-Demo, A.; Cara, A.; Gallo, R.C.; Lusso, P. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat. Med. 1996, 2, 1244–1247. [Google Scholar] [CrossRef]
- Donzella, G.A.; Schols, D.; Lin, S.W.; Este, J.A.; Nagashima, K.A.; Maddon, P.J.; Allaway, G.P.; Sakmar, T.P.; Henson, G.; De Clercq, E.; Moore, J.P. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 1998, 4, 72–77. [Google Scholar]
- Hendrix, C.W.; Collier, A.C.; Lederman, M.M.; Schols, D.; Pollard, R.B.; Brown, S.; Jackson, J.B.; Coombs, R.W.; Glesby, M.J.; Flexner, C.W.; Bridger, G.J.; Badel, K.; MacFarland, R.T.; Henson, G.W.; Calandra, G.; the AMD 3100 HIV Study Group. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J. Acquir. Defic. Syndr. 2004, 37, 1253–1262. [Google Scholar] [CrossRef]
- Hatse, S.; Princen, K.; Bridger, G.; De Clercq, E.; Schols, D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002, 527, 255–262. [Google Scholar] [CrossRef]
- Fricker, S.P.; Anastassov, V.; Cox, J.; Darkes, M.C.; Grujic, O.; Idzan, S.R.; Labrecque, J.; Lau, G.; Mosi, R.M.; Nelson, K.L.; Qin, L.; Santucci, Z.; Wong, R.S.Y. Characterization of the molecular pharmacology of AMD3100: a specific antagonist of the G-protein coupled chemokine receptor, CXCR4. Biochem. Pharmacol. 2006, 72, 588–596. [Google Scholar] [CrossRef]
- Mastrolorenzo, A.; Scozzafava, A.; Supuran, C.T. Small molecule antagonists of chemokine receptors as emerging anti-HIV agents. Expert Opin. Ther. Pat. 2001, 11, 1245–1252. [Google Scholar] [CrossRef]
- Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Non-peptidic chemokine receptors antagonists as emerging anti-HIV agents. J. Enz. Inhib. Med. Chem. 2002, 17, 69–76. [Google Scholar] [CrossRef]
- Rusconi, S.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. New advances in HIV entry inhibitors development. Curr. Drug Targets – Infect. Dis. 2004, 4, 339–355. [Google Scholar] [CrossRef]
- Rusconi, S.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. An update in the development of HIV entry inhibitors. Curr. Top. Med. Chem. 2007, 7, 1273–1289. [Google Scholar] [CrossRef]
- Fricker, S.P. A novel CXCR4 antagonist for hematopoietic stem cell mobilization. Expert Opin. Investig. Drugs 2008, 17, 1749–1760. [Google Scholar] [CrossRef]
- Hatse, S.; Princen, K.; De Clercq, E.; Rosenkilde, M.M.; Schwartz, T.W.; Hernandez-Abad, P.E.; Skerlj, R.T.; Bridger, G.J.; Schols, D. AMD3465, a monomacrocyclic CXCR4 antagonist and potent HIV entry inhibitor. Biochem. Pharmacol. 2005, 70, 752–761. [Google Scholar] [CrossRef]
- Rusconi, S.; Merrill, D.P.; La Seta Catamancio, S.; Citterio, P.; Bulgheroni, E.; Croce, F.; Chou, T.C.; Yang, O.O.; Herrmann, S.H.; Galli, M.; Hirsch, M.S. In vitro inhibition of HIV-1 by Met-SDF-1β alone or in combination with antiretroviral agents. Antiviral Ther. 2000, 5, 199–204. [Google Scholar]
- Tremblay, C.L.; Giguel, F.; Kollman, C.; Guan, Y.; Chou, T.C.; Baroudy, B.M.; Hirsch, M.S. Anti-human immunodeficiency virus interactions of SCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob. Agents Chemother. 2002, 46, 1336–1339. [Google Scholar] [CrossRef]
- Tremblay, C.L.; Giguel, F.; Guan, Y.; Chou, T.C.; Takashima, K.; Hirsch., M.S. TAK-220, a novel small-molecule CCR5 antagonist, has favorable anti-human immunodeficiency virus interactions with other antiretrovirals in vitro. Antimicrob. Agents Chemother. 2005, 49, 3483–3485. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Sorensen, M.; Fung, M.; Schooley, R.T. Synergistic in vitro antiretroviral activity of a humanized monoclonal anti-CD4 antibody (TNX-355) and enfuvirtide (T-20). Antimicrob. Agents Chemother. 2006, 50, 2231–2233. [Google Scholar] [CrossRef]
- Bazzicalupi, C.; Bencini, A.; Fusi, V.; Giorgi, C.; Paoletti, P.; Valtancoli, B. Lead complexation by novel phenanthroline-containing macrocycles. J. Chem. Soc., Dalton Trans. 1999, 393–399. [Google Scholar]
- Bazzicalupi, C.; Bencini, A.; Ciattini, S.; Giorgi, C.; Masotti, A.; Paoletti, P.; Valtancoli, B.; Navon, N.; Meyerstein, D. Copper-(II) and -(I) co-ordination by hexa-amine ligands of different rigidities. A thermodynamic, structural and electrochemical investigation. J. Chem. Soc. Dalton Trans. 2000, 2383–2391. [Google Scholar]
- Bazzicalupi, C.; Bellusci, A.; Bencini, A.; Berni, E.; Bianchi, A.; Ciattini, S.; Giorgi, C.; Valtancoli, B. A new dipyridine-containing cryptand for both proton and Cu(II) encapsulation. A solution and solid state study. J. Chem. Soc. Dalton Trans. 2002, 2151–2157. [Google Scholar]
- Bencini, A.; Bianchi, A.; Lodeiro, C.; Masotti, A.; Parola, J.A.; Melo, J.S.; Pina, F.; Valtancoli, B. A novel fluorescent chemosensor exhibiting exciplex emission. An example of an elementary molecular machine driven by pH and by light. Chem. Commun. 2000, 1639–1640. [Google Scholar]
- Chandler, C.J.; Deady, L.W.; Reiss, J.A. Synthesis of some 2,9-disubstituted-1,10-phenanthrolines. J. Heterocycl. Chem. 1981, 18, 599–605. [Google Scholar] [CrossRef]
- Bencini, A.; Burguete, M.I.; Garcia-España, E.; Luis, S.V.; Miravet, J.F.; Soriano, C. An Efficient Synthesis of Polyaza[n]paracyclophanes. J. Org. Chem. 1993, 58, 4749–4753. [Google Scholar] [CrossRef]
- Motekaitis, R.J.; Martell, A.E.; Murase, I. Cascade halide binding by multiprotonated 7,19,30-trioxa-1,4,10,13,16,22,27,33octaazabicyclo[11.11.11]pentatriacontane (BISTREN) and copper(II) BISTREN cryptates. Inorg. Chem. 1986, 25, 938–944. [Google Scholar] [CrossRef]
- Arago, J.; Bencini, A.; Bianchi, A.; Garcia-España, E.; Micheloni, M.; Paoletti, P.; Ramirez, J.A.; Paoli, P. Interaction of “Long” Open-Chain Polyazaalkanes with Hydrogen and Copper(II) Ions. Inorg. Chem. 1991, 30, 1843–1847. [Google Scholar] [CrossRef]
- Bencini, A.; Bianchi, A.; Borselli, A.; Chimichi, S.; Ciampolini, M.; Dapporto, P.; Micheloni, M.; Nardi, N.; Paoli, P.; Valtancoli, B. Selective lithium encapsulation in aqueous solution by the new cage 4,l0-dimethyl-1,4,7,10,15-pentaazabicyclo[5.5.5]heptadecane (L) Synthesis, characterization, and structural aspects. Crystal structures of [LiL][ClO4] and [CuL]Br2.3H2O. Inorg. Chem. 1990, 29, 3282–3286. [Google Scholar] [CrossRef]
- Björndal, Å.; Deng, H.; Jansson, M.; Fiore, J.R.; Colognesi, C.; Karlsson, A.; Albert, J.; Scarlatti, G.; Littman, D.R.; Fenyö, E.M. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 1997, 71, 7478–7487. [Google Scholar]
- Rusconi, S.; La Seta Catamancio, S.; Citterio, P.; Bulgheroni, E.; Croce, F.; Herrmann, S.H.; Offord, R.E.; Galli, M.; Hirsch, M.S. Combinations of CCR5 and CXCR4 inhibitors in therapy of human immunodeficiency virus type 1 infection: in vitro studies of mixed virus infections. J. Virol. 2000, 74, 9328–9332. [Google Scholar]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, T.C. The median-effect principle and the combination index for quantitation of synergism and antagonism. In Synergism and Antagonism in Chemotherapy; Chou, T.C., Rideout, D.C., Eds.; Academic Press, Inc.: San Diego, CA, USA, 1991; pp. 61–102. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rusconi, S.; Cicero, M.L.; Viganò, O.; Sirianni, F.; Bulgheroni, E.; Ferramosca, S.; Bencini, A.; Bianchi, A.; Ruiz, L.; Cabrera, C.; et al. New Macrocyclic Amines Showing Activity as HIV Entry Inhibitors Against Wild Type and Multi-Drug Resistant Viruses. Molecules 2009, 14, 1927-1937. https://doi.org/10.3390/molecules14051927
Rusconi S, Cicero ML, Viganò O, Sirianni F, Bulgheroni E, Ferramosca S, Bencini A, Bianchi A, Ruiz L, Cabrera C, et al. New Macrocyclic Amines Showing Activity as HIV Entry Inhibitors Against Wild Type and Multi-Drug Resistant Viruses. Molecules. 2009; 14(5):1927-1937. https://doi.org/10.3390/molecules14051927
Chicago/Turabian StyleRusconi, Stefano, Mirko Lo Cicero, Ottavia Viganò, Francesca Sirianni, Elisabetta Bulgheroni, Stefania Ferramosca, Andrea Bencini, Antonio Bianchi, Lidia Ruiz, Cecilia Cabrera, and et al. 2009. "New Macrocyclic Amines Showing Activity as HIV Entry Inhibitors Against Wild Type and Multi-Drug Resistant Viruses" Molecules 14, no. 5: 1927-1937. https://doi.org/10.3390/molecules14051927
APA StyleRusconi, S., Cicero, M. L., Viganò, O., Sirianni, F., Bulgheroni, E., Ferramosca, S., Bencini, A., Bianchi, A., Ruiz, L., Cabrera, C., Martinez-Picado, J., Supuran, C. T., & Galli, M. (2009). New Macrocyclic Amines Showing Activity as HIV Entry Inhibitors Against Wild Type and Multi-Drug Resistant Viruses. Molecules, 14(5), 1927-1937. https://doi.org/10.3390/molecules14051927