Mouse Models Targeting Selenocysteine tRNA Expression for Elucidating the Role of Selenoproteins in Health and Development

Abstract
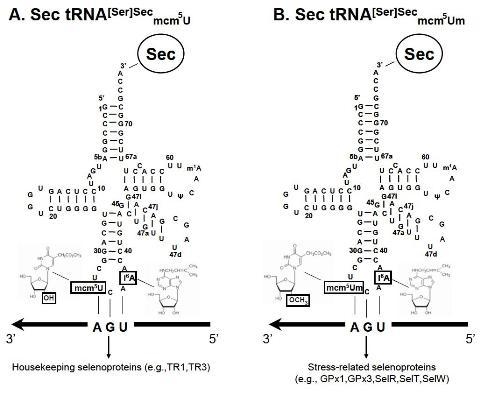
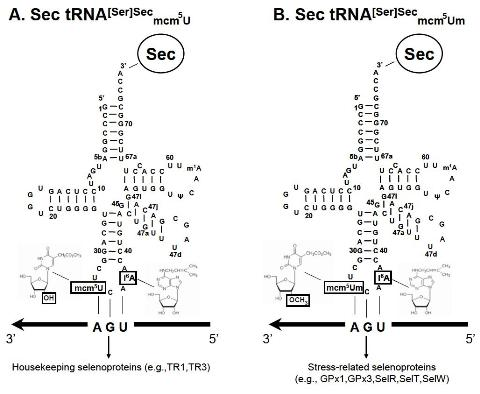
:
1. Introduction
2. Mouse Models Involving the Sec tRNA[Ser]Sec Gene (Trsp)
2.1. Trsp transgenic mouse models


{kind=link}
{kind=link}
| Transgene Numberb | Model Description | Major Findings | Reference |
|---|---|---|---|
| 2-4, 8-16, 20-40 | Mice encode a mutant G37 transgene in all tissues and organs. | Levels of stress-related selenoproteins decreased in a protein and tissue specific manner in mice expressing a mutant G37 tRNA[Ser]Sec isoform that also lacks Um34. GPx1 and TR3 were the most and least affected selenoproteins, while selenoprotein expression was most and least affected in the liver and testes, respectively. First transgenic mouse generated encoding a tRNA transgene. | [9] |
| 40 | Mice encode a mutant G37 transgene in all tissues and organs. | Enhanced skeletal muscle adaptation after exercise enhanced growth in the G37 mice that was completely blocked by inhibition of the mammalian target of rapamycin (mTOR) pathway. Muscles of transgenic mice exhibited increased site-specific phosphorylation on both Akt and p70 ribosomal S6 kinase before ablation. | [72] |
| 40 | Mice encode a mutant G37 transgene in all tissues and organs. Colon is targeted with azoxymethane. | Mice had more azoxymethane-induced aberrant crypt formation (a preneoplastic lesion for colon cancer). First demonstration that selenoproteins reduce colon cancer incidence. | [40] |
| 20 | Mice encode a mutant G37 transgene in all tissues and organs and a prostate cancer driving C3(1)/Tag transgene. | Mutant mice exhibited accelerated development of lesions associated with prostate cancer progression, implicating selenoproteins in cancer risk and raising the possibility that Se prevents cancer by modulating the levels of stress-related selenoproteins. First demonstration that selenoproteins reduce prostate cancer incidence. | [42] |
| 40 | Mice encode a mutant G37 transgene in all tissues and organs. | Mutant mice showed higher micronuclei formation than control mice in erythrocytes following exposure to X-rays. | [73] |
| 40 | Mice encode a mutant G37 transgene in all tissues and organs. Lung is targeted by administration of influenza virus. | At day 2 p.i., chemokine levels were greater in the G37 mice compared with wild type mice. Additionally, IFN-γ was higher at day 7 p.i. in the G37 mice and viral clearance slower. Despite these immune system changes, lung pathology was similar in G37 and wild type mice. | [74] |
2.2. Trsp conditional knockout mouse models
| Cre Promoter | Targeted Organ or Tissue | Major Findings | Reference |
|---|---|---|---|
| MMTV-Cre; Wap-Cre | Mammary gland | First description of the Trsp conditional knockout mouse. | [11] |
| Alb-Cre | Liver | Death between 1 and 3 months of age due to severe hepatocellular degeneration and necrosis. Elevated GST levels [12]. Brain Se levels are maintained in the absence of liver-derived plasma SePP [75]. Hepatic Dio1 is not essential to maintain plasma thyroid hormone levels [76]. Selenoproteins have a role in proper liver function. | [12,75,76] |
| TieTek2-Cre | Endothelial cell | 14.5 dpc embryos were smaller in size, more fragile, had a poorly developed vascular system, underdeveloped limbs and tails and heads. Selenoproteins have a role in endothelial cell function. | [53] |
| MCK-Cre | Heart and skeletal muscle | Died from acute myocardial failure day 12 after birth. Selenoproteins have a role in preventing heart disease. | [53] |
| LysM-Cre | Macrophage | Elevated oxidative stress and transcriptional induction of cytoprotective antioxidant and detoxification enzyme genes. Accumulation of ROS levels and impaired invasiveness. Altered expression of several extracellular matrix and fibrosis-associated genes. Selenoproteins have a role in immune function. | [46]; Carlson et al.a |
| NPHS2-Cre | Kidney | Loss of podocyte selenoproteins does not lead to increased oxidative stress or worsening nephropathy. | [77] |
| LCK-Cre | T cells | Decreased pools of mature T cells and a defect in T cell-dependent antibody responses. Antioxidant hyperproduction and thereby suppression of T cell proliferation in response to T cell receptor stimulation. Selenoproteins have a role in immune function. | [45] |
| Tα1 antigen-Cre | Neuron specific | Enhanced neuronal excitation followed by massive neurodegeneration of the hippocampus. Cerebellar hypoplasia was associated with degeneration of Purkinje and granule cells. Cerebellar interneurons were essentially absent. Selenoproteins have a role in neuronal function. | Schweizer et al.a |
| Col2a1-Cre | Osteo-chondroprogenitor | Post-natal growth retardation, chondrodyplasia, chondronecrosis and delayed skeletal ossification characteristic of Kashin-Beck disease. First model for Kashin-Beck disease. | [48] |
| K14-Cre | Skin | Runt phenotype, premature death, alopecia along with a flaky and fragile skin, epidermal hyperplasia with disturbed hair cycle and an early regression of hair follicles. Selenoproteins have a role in skin and hair follicle development. | Sengupta et al.a |
2.2.1. Mouse models relating to human disease
2.3. Trsp transgenic/standard or transgenic/conditional knockout mouse models
| Transgene and Numbera | Model Description | Major Findings | Reference |
|---|---|---|---|
| G37 (40) | All tissues lack a wild type Trsp gene and are rescued with mutant G37 transgenes. | The absence of Um34 plays a major role in the expression of stress-related selenoproteins, but not housekeeping selenoproteins. | [10] |
| A34 (2); G37 (2, 16) | Trsp is removed in liver and the resulting mouse encodes either mutant A34 or G37 transgenes. | Both mutant tRNAs lacked Um34, and both supported expression of housekeeping selenoproteins (e.g., TR1) in liver, but not stress-related proteins (e.g., GPx 1). Um34 is responsible for synthesis of a select group of selenoproteins, the stress-related selenoproteins, rather than the entire selenoprotein population. | [28] |
| A34 (2); G37 (2, 16) | Trsp is removed in liver and the resulting mouse encodes either mutant A34 or G37 transgenes. | In Trsp mutant mouse lines, the expression of ApoE, as well as genes involved in cholesterol biosynthesis, metabolism and transport were similar to those observed in wild type mice indicating for the first time that housekeeping selenoproteins have a role in regulating lipoprotein biosynthesis and metabolism. | [55] |
| A34 (2); G37 (2, 16) | Trsp is removed in liver and the resulting mouse encodes either mutant A34 or G37 transgenes. | The loss of selenoproteins in liver was compensated for by an enhanced expression of several phase II response genes and their corresponding gene products. The replacement of selenoprotein synthesis in mice carrying mutant Trsp transgenes led to normal expression of phase II response genes. Provides evidence for a functional link between housekeeping selenoproteins and phase II enzymes. | [78] |
2.4. Other mouse models involving Trsp
3. Conclusions
Acknowledgements
- Sample Availability: Mouse tissue samples are available from the authors.
References
- Hatfield, D.L.; Berry, M.J.; Gladyshev, V.N. Selenium: Its Molecular Biology and Role in Human Health; Springer Science+Business Media, LLC: New York, NY, USA, 2006. [Google Scholar]
- Birringer, M.; Pilawa, S.; Flohe, L. Trends in selenium biochemistry. Nat. Prod. Rep. 2002, 19, 693–718. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Gladyshev, V.N. How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef]
- Stadtman, T.C. Discoveries of vitamin B12 and selenium enzymes. Annu. Rev. Biochem. 2002, 71, 1–16. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Choi, I.S.; Ohama, T.; Jung, J.E.; Diamond, A.M. Selenocysteine tRNA(Ser)sec isoacceptors as central components in selenoprotein biosynthesis in eukaryotes. In Selenium in Biology and Human Health; Burk, R.F., Ed.; Springer-Verlag: New York, NY, USA, 1994; pp. 25–44. [Google Scholar]
- Xu, X.M.; Carlson, B.A.; Mix, H.; Zhang, Y.; Saira, K.; Glass, R.S.; Berry, M.J.; Gladyshev, V.N.; Hatfield, D.L. Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol. 2006, 5, e4. [Google Scholar]
- Yuan, J.; Palioura, S.; Salazar, J.C.; Su, D.; O'Donoghue, P.; Hohn, M.J.; Cardoso, A.M.; Whitman, W.B.; Soll, D. RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc. Natl. Acad. Sci. USA 2006, 103, 18923–18927. [Google Scholar]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar]
- Moustafa, M.E.; Carlson, B.A.; El-Saadani, M.A.; Kryukov, G.V.; Sun, Q.A.; Harney, J.W.; Hill, K.E.; Combs, G.F.; Feigenbaum, L.; Mansur, D.B.; Burk, R.F.; Berry, M.J.; Diamond, A.M.; Lee, B.J.; Gladyshev, V.N.; Hatfield, D.L. Selective inhibition of selenocysteine tRNA maturation and selenoprotein synthesis in transgenic mice expressing isopentenyladenosine-deficient selenocysteine tRNA. Mol. Cell. Biol. 2001, 21, 3840–3852. [Google Scholar]
- Carlson, B.A.; Xu, X.M.; Gladyshev, V.N.; Hatfield, D.L. Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine tRNA. J. Biol. Chem. 2005, 280, 5542–5548. [Google Scholar]
- Kumaraswamy, E.; Carlson, B.A.; Morgan, F.; Miyoshi, K.; Robinson, G.W.; Su, D.; Wang, S.; Southon, E.; Tessarollo, L.; Lee, B.J.; Gladyshev, V.N.; Hennighausen, L.; Hatfield, D.L. Selective removal of the selenocysteine tRNA [Ser]Sec gene (Trsp) in mouse mammary epithelium. Mol. Cell. Biol. 2003, 23, 1477–1488. [Google Scholar]
- Carlson, B.A.; Novoselov, S.V.; Kumaraswamy, E.; Lee, B.J.; Anver, M.R.; Gladyshev, V.N.; Hatfield, D.L. Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J. Biol. Chem. 2004, 279, 8011–8017. [Google Scholar]
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 1997, 272, 16644–16651. [Google Scholar]
- Esworthy, R.S.; Mann, J.R.; Sam, M.; Chu, F.F. Low glutathione peroxidase activity in Gpx1 knockout mice protects jejunum crypts from gamma-irradiation damage. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G426–436. [Google Scholar]
- Esworthy, R.S.; Aranda, R.; Martin, M.G.; Doroshow, J.H.; Binder, S.W.; Chu, F.F. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G848–855. [Google Scholar]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Jakupoglu, C.; Przemeck, G.K.; Schneider, M.; Moreno, S.G.; Mayr, N.; Hatzopoulos, A.K.; de Angelis, M.H.; Wurst, W.; Bornkamm, G.W.; Brielmeier, M.; Conrad, M. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol. Cell. Biol. 2005, 25, 1980–1988. [Google Scholar]
- Conrad, M.; Jakupoglu, C.; Moreno, S.G.; Lippl, S.; Banjac, A.; Schneider, M.; Beck, H.; Hatzopoulos, A.K.; Just, U.; Sinowatz, F.; Schmahl, W.; Chien, K.R.; Wurst, W.; Bornkamm, G.W.; Brielmeier, M. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell. Biol. 2004, 24, 9414–9423. [Google Scholar]
- Schneider, M.J.; Fiering, S.N.; Thai, B.; Wu, S.Y.; St Germain, E.; Parlow, A.F.; St Germain, D.L.; Galton, V.A. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology 2006, 147, 580–589. [Google Scholar]
- Schneider, M.J.; Fiering, S.N.; Pallud, S.E.; Parlow, A.F.; St Germain, D.L.; Galton, V.A. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol. Endocrinol. 2001, 15, 2137–2148. [Google Scholar] [CrossRef]
- Galton, V.A.; Schneider, M.J.; Clark, A.S.; St Germain, D.L. Life without thyroxine to 3,5,3'-triiodothyronine conversion: studies in mice devoid of the 5'-deiodinases. Endocrinology 2009, 150, 2957–2963. [Google Scholar] [CrossRef]
- Hernandez, A.; Martinez, M.E.; Fiering, S.; Galton, V.A.; St Germain, D. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J. Clin. Invest. 2006, 116, 476–484. [Google Scholar] [CrossRef]
- Schomburg, L.; Schweizer, U.; Holtmann, B.; Flohe, L.; Sendtner, M.; Kohrle, J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 2003, 370, 397–402. [Google Scholar] [CrossRef]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Burk, R.F. Neurological dysfunction occurs in mice with targeted deletion of the selenoprotein P gene. J. Nutr. 2004, 134, 157–161. [Google Scholar]
- Fomenko, D.E.; Novoselov, S.V.; Natarajan, S.K.; Lee, B.C.; Koc, A.; Carlson, B.A.; Lee, T.H.; Kim, H.Y.; Hatfield, D.L.; Gladyshev, V.N. MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J. Biol. Chem. 2009, 284, 5986–5993. [Google Scholar]
- Conrad, M.; Schweizer, U. Unveiling the molecular mechanisms behind selenium-related diseases through knockout mouse studies. Antioxid. Redox Signal. 2009, in press. [Google Scholar]
- Bosl, M.R.; Takaku, K.; Oshima, M.; Nishimura, S.; Taketo, M.M. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc. Natl. Acad. Sci. USA 1997, 94, 5531–5534. [Google Scholar]
- Carlson, B.A.; Moustafa, M.E.; Sengupta, A.; Schweizer, U.; Shrimali, R.; Rao, M.; Zhong, N.; Wang, S.; Feigenbaum, L.; Lee, B.J.; Gladyshev, V.N.; Hatfield, D.L. Selective restoration of the selenoprotein population in a mouse hepatocyte selenoproteinless background with different mutant selenocysteine tRNAs lacking Um34. J. Biol. Chem. 2007, 282, 32591–32602. [Google Scholar]
- Diamond, A.M.; Choi, I.S.; Crain, P.F.; Hashizume, T.; Pomerantz, S.C.; Cruz, R.; Steer, C.J.; Hill, K.E.; Burk, R.F.; McCloskey, J.A.; et al. Dietary selenium affects methylation of the wobble nucleoside in the anticodon of selenocysteine tRNA([Ser]Sec). J. Biol. Chem. 1993, 268, 14215–14223. [Google Scholar]
- Choi, I.S.; Diamond, A.M.; Crain, P.F.; Kolker, J.D.; McCloskey, J.A.; Hatfield, D.L. Reconstitution of the biosynthetic pathway of selenocysteine tRNAs in Xenopus oocytes. Biochemistry 1994, 33, 601–605. [Google Scholar]
- Kim, L.K.; Matsufuji, T.; Matsufuji, S.; Carlson, B.A.; Kim, S.S.; Hatfield, D.L.; Lee, B.J. Methylation of the ribosyl moiety at position 34 of selenocysteine tRNA[Ser]Sec is governed by both primary and tertiary structure. RNA 2000, 6, 1306–1315. [Google Scholar] [CrossRef]
- Hatfield, D.; Lee, B.J.; Hampton, L.; Diamond, A.M. Selenium induces changes in the selenocysteine tRNA[Ser]Sec population in mammalian cells. Nucleic Acids Res. 1991, 19, 939–943. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Carlson, B.A.; Xu, X.M.; Mix, H.; Gladyshev, V.N. Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 97–142. [Google Scholar] [CrossRef]
- Sun, X.; Li, X.; Moriarty, P.M.; Henics, T.; LaDuca, J.P.; Maquat, L.E. Nonsense-mediated decay of mRNA for the selenoprotein phospholipid hydroperoxide glutathione peroxidase is detectable in cultured cells but masked or inhibited in rat tissues. Mol. Biol. Cell 2001, 12, 1009–1017. [Google Scholar]
- Weiss, S.L.; Sunde, R.A. Cis-acting elements are required for selenium regulation of glutathione peroxidase-1 mRNA levels. RNA 1998, 4, 816–827. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R. Selenium compounds and selenoproteins in cancer. Chem. Biodivers. 2008, 5, 389–395. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium in cancer prevention: a review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef]
- Ip, C.; Hayes, C.; Budnick, R.M.; Ganther, H.E. Chemical form of selenium, critical metabolites, and cancer prevention. Cancer Res. 1991, 51, 595–600. [Google Scholar]
- Lu, J.; Jiang, C. Selenium and cancer chemoprevention: hypotheses integrating the actions of selenoproteins and selenium metabolites in epithelial and non-epithelial target cells. Antioxid. Redox Signal. 2005, 7, 1715–1727. [Google Scholar] [CrossRef]
- Irons, R.; Carlson, B.A.; Hatfield, D.L.; Davis, C.D. Both selenoproteins and low molecular weight selenocompounds reduce colon cancer risk in mice with genetically impaired selenoprotein expression. J. Nutr. 2006, 136, 1311–1317. [Google Scholar]
- Kipp, A.; Banning, A.; Schothorst, E.M.v.; Méplan, C.; Schomburg, L.; Evelo, C.; Coort, S.; Gaj, S.; Keijer, J.; Hesketh, J.; Brigelius-Flohé, R. Four selenoproteins, protein biosynthesis, and Wnt signalling are particularly sensitive to selenium intake in mouse colon. Mol. Nutr. Food Res. 2009, in press. [Google Scholar]
- Diwadkar-Navsariwala, V.; Prins, G.S.; Swanson, S.M.; Birch, L.A.; Ray, V.H.; Hedayat, S.; Lantvit, D.L.; Diamond, A.M. Selenoprotein deficiency accelerates prostate carcinogenesis in a transgenic model. Proc. Natl. Acad. Sci. USA 2006, 103, 8179–8184. [Google Scholar]
- Hoffmann, P.R. Mechanisms by which selenium influences immune responses. Arch. Immunol. Ther. Exp. (Warsz) 2007, 55, 289–297. [Google Scholar] [CrossRef]
- Hoffmann, P.R.; Berry, M.J. The influence of selenium on immune responses. Mol. Nutr. Food Res. 2008, 52, 1273–1280. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Irons, R.D.; Carlson, B.A.; Sano, Y.; Gladyshev, V.N.; Park, J.M.; Hatfield, D.L. Selenoproteins mediate T cell immunity through an antioxidant mechanism. J. Biol. Chem. 2008, 283, 20181–20185. [Google Scholar]
- Suzuki, T.; Kelly, V.P.; Motohashi, H.; Nakajima, O.; Takahashi, S.; Nishimura, S.; Yamamoto, M. Deletion of the selenocysteine tRNA gene in macrophages and liver results in compensatory gene induction of cytoprotective enzymes by Nrf2. J. Biol. Chem. 2008, 283, 2021–2030. [Google Scholar]
- Kolsteren, P. Kashin-Beck disease. Ann. Soc. Belg. Med. Trop. 1992, 72, 81–91. [Google Scholar]
- Downey, C.M.; Horton, C.R.; Carlson, B.A.; Parsons, T.E.; Hatfield, D.L.; Hallgrímsson, B.; Jirik, F.R. Osteo-chondroprogenitor-specific deletion of the selenocysteine tRNA gene, Trsp, leads to chondronecrosis and abnormal skeletal development: a putative model for Kashin-Beck disease. PLoS Genet. 2009, in press. [Google Scholar]
- Ge, K.; Xue, A.; Bai, J.; Wang, S. Keshan disease-an endemic cardiomyopathy in China. Virchows Arch. A Pathol. Anat. Histopathol. 1983, 401, 1–15. [Google Scholar] [CrossRef]
- Cheng, Y.Y.; Qian, P.C. The effect of selenium-fortified table salt in the prevention of Keshan disease on a population of 1.05 million. Biomed. Environ. Sci. 1990, 3, 422–428. [Google Scholar]
- Beck, M.A.; Levander, O.A.; Handy, J. Selenium deficiency and viral infection. J. Nutr. 2003, 133, 1463–1467. [Google Scholar]
- Gao, J.; Xiong, Y.; Ho, Y.S.; Liu, X.; Chua, C.C.; Xu, X.; Wang, H.; Hamdy, R.; Chua, B.H. Glutathione peroxidase 1-deficient mice are more susceptible to doxorubicin-induced cardiotoxicity. Biochim. Biophys. Acta 2008, 1783, 2020–2029. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Weaver, J.A.; Miller, G.F.; Starost, M.F.; Carlson, B.A.; Novoselov, S.V.; Kumaraswamy, E.; Gladyshev, V.N.; Hatfield, D.L. Selenoprotein expression is essential in endothelial cell development and cardiac muscle function. Neuromuscul. Disord. 2007, 17, 135–142. [Google Scholar]
- Moustafa, M.E.; El-Saadani, M.A.; Kandeel, K.M.; Mansur, D.B.; Lee, B.J.; Hatfield, D.L.; Diamond, A.M. Overproduction of selenocysteine tRNA in Chinese hamster ovary cells following transfection of the mouse tRNA[Ser]Sec gene. RNA 1998, 4, 1436–1443. [Google Scholar] [CrossRef]
- Sengupta, A.; Carlson, B.A.; Hoffmann, V.J.; Gladyshev, V.N.; Hatfield, D.L. Loss of housekeeping selenoprotein expression in mouse liver modulates lipoprotein metabolism. Biochem. Biophys. Res. Commun. 2008, 365, 446–452. [Google Scholar] [CrossRef]
- Kelly, V.P.; Suzuki, T.; Nakajima, O.; Arai, T.; Tamai, Y.; Takahashi, S.; Nishimura, S.; Yamamoto, M. The distal sequence element of the selenocysteine tRNA gene is a tissue-dependent enhancer essential for mouse embryogenesis. Mol. Cell. Biol. 2005, 25, 3658–3669. [Google Scholar]
- Carlson, B.A.; Schweizer, U.; Perella, C.; Shrimali, R.K.; Feigenbaum, L.; Shen, L.; Speransky, S.; Floss, T.; Jeong, S.J.; Watts, J.; Hoffmann, V.; Combs, G.F.; Gladyshev, V.N.; Hatfield, D.L. The selenocysteine tRNA STAF-binding region is essential for adequate selenocysteine tRNA status, selenoprotein expression and early age survival of mice. Biochem. J. 2009, 418, 61–71. [Google Scholar] [CrossRef]
- Lee, B.J.; de la Pena, P.; Tobian, J.A.; Zasloff, M.; Hatfield, D. Unique pathway of expression of an opal suppressor phosphoserine tRNA. Proc. Natl. Acad. Sci. USA 1987, 84, 6384–6388. [Google Scholar] [CrossRef]
- Lee, B.J.; Kang, S.G.; Hatfield, D. Transcription of Xenopus selenocysteine tRNA Ser (formerly designated opal suppressor phosphoserine tRNA) gene is directed by multiple 5'-extragenic regulatory elements. J. Biol. Chem. 1989, 264, 9696–9702. [Google Scholar]
- Myslinski, E.; Krol, A.; Carbon, P. Optimal tRNA((Ser)Sec) gene activity requires an upstream SPH motif. Nucleic Acids Res. 1992, 20, 203–209. [Google Scholar] [CrossRef]
- Park, J.M.; Choi, I.S.; Kang, S.G.; Lee, J.Y.; Hatfield, D.L.; Lee, B.J. Upstream promoter elements are sufficient for selenocysteine tRNA[Ser]Sec gene transcription and to determine the transcription start point. Gene 1995, 162, 13–19. [Google Scholar] [CrossRef]
- Adachi, K.; Katsuyama, M.; Song, S.; Oka, T. Genomic organization, chromosomal mapping and promoter analysis of the mouse selenocysteine tRNA gene transcription-activating factor (mStaf) gene. Biochem. J. 2000, 346 (Pt 1), 45–51. [Google Scholar] [CrossRef]
- Adachi, K.; Saito, H.; Tanaka, T.; Oka, T. Molecular cloning and characterization of the murine staf cDNA encoding a transcription activating factor for the selenocysteine tRNA gene in mouse mammary gland. J. Biol. Chem. 1998, 273, 8598–8606. [Google Scholar] [CrossRef]
- Adachi, K.; Tanaka, T.; Saito, H.; Oka, T. Hormonal induction of mouse selenocysteine transfer ribonucleic acid (tRNA) gene transcription-activating factor and its functional importance in the selenocysteine tRNA gene transcription in mouse mammary gland. Endocrinology 1999, 140, 618–623. [Google Scholar] [CrossRef]
- Schuster, C.; Myslinski, E.; Krol, A.; Carbon, P. Staf, a novel zinc finger protein that activates the RNA polymerase III promoter of the selenocysteine tRNA gene. EMBO J. 1995, 14, 3777–3787. [Google Scholar]
- Barski, O.A.; Papusha, V.Z.; Kunkel, G.R.; Gabbay, K.H. Regulation of aldehyde reductase expression by STAF and CHOP. Genomics 2004, 83, 119–129. [Google Scholar]
- Schaub, M.; Krol, A.; Carbon, P. Flexible zinc finger requirement for binding of the transcriptional activator staf to U6 small nuclear RNA and tRNA(Sec) promoters. J. Biol. Chem. 1999, 274, 24241–24249. [Google Scholar]
- Schaub, M.; Myslinski, E.; Krol, A.; Carbon, P. Maximization of selenocysteine tRNA and U6 small nuclear RNA transcriptional activation achieved by flexible utilization of a Staf zinc finger. J. Biol. Chem. 1999, 274, 25042–25050. [Google Scholar]
- Schaub, M.; Myslinski, E.; Schuster, C.; Krol, A.; Carbon, P. Staf, a promiscuous activator for enhanced transcription by RNA polymerases II and III. EMBO J. 1997, 16, 173–181. [Google Scholar] [CrossRef]
- Hatfield, D.; Gladyshev, V.; Park, J.M.; Park, S.I.; Chittum, H.; Huh, J.; Carlson, B.; Kim, M.; Moustafa, M.; Lee, B.J. Biosynthesis of selenocysteine and its incorporation into protein as the 21st amino acid. In Comprehensive Natural Products Chemistry; Kelly, J.W., Ed.; Elsevier Science: Oxford, U.K, 1999; Volume 4, pp. 353–380. [Google Scholar]
- Schweizer, U.; Michaelis, M.; Kohrle, J.; Schomburg, L. Efficient selenium transfer from mother to offspring in selenoprotein-P-deficient mice enables dose-dependent rescue of phenotypes associated with selenium deficiency. Biochem. J. 2004, 378, 21–26. [Google Scholar] [CrossRef]
- Hornberger, T.A.; McLoughlin, T.J.; Leszczynski, J.K.; Armstrong, D.D.; Jameson, R.R.; Bowen, P.E.; Hwang, E.S.; Hou, H.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Diamond, A.M.; Esser, K.A. Selenoprotein-deficient transgenic mice exhibit enhanced exercise-induced muscle growth. J. Nutr. 2003, 133, 3091–3097. [Google Scholar]
- Baliga, M.S.; Diwadkar-Navsariwala, V.; Koh, T.; Fayad, R.; Fantuzzi, G.; Diamond, A.M. Selenoprotein deficiency enhances radiation-induced micronuclei formation. Mol. Nutr. Food Res. 2008, 52, 1300–1304. [Google Scholar] [CrossRef]
- Sheridan, P.A.; Zhong, N.; Carlson, B.A.; Perella, C.M.; Hatfield, D.L.; Beck, M.A. Decreased selenoprotein expression alters the immune response during influenza virus infection in mice. J. Nutr. 2007, 137, 1466–1471. [Google Scholar]
- Schweizer, U.; Streckfuss, F.; Pelt, P.; Carlson, B.A.; Hatfield, D.L.; Köhrle, J.; Schomburg, L. Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem. J. 2005, 386, 221–226. [Google Scholar] [CrossRef]
- Streckfuss, F.; Hamann, I.; Schomburg, L.; Michaelis, M.; Sapin, R.; Klein, M.O.; Köhrle, J.; Schweizer, U. Hepatic deiodinase activity is dispensable for the maintenance of normal circulating thyroid hormone levels in mice. Biochem. Biophys. Res. Commun. 2005, 337, 739–745. [Google Scholar] [CrossRef]
- Blauwkamp, M.N.; Yu, J.; Schin, M.A.; Burke, K.A.; Berry, M.J.; Carlson, B.A.; Brosius, F.C., 3rd; Koenig, R.J. Podocyte specific knock out of selenoproteins does not enhance nephropathy in streptozotocin diabetic C57BL/6 mice. BMC Nephrol. 2008, 9, 7. [Google Scholar] [CrossRef]
- Sengupta, A.; Carlson, B.A.; Weaver, J.A.; Novoselov, S.V.; Fomenko, D.E.; Gladyshev, V.N.; Hatfield, D.L. A functional link between housekeeping selenoproteins and phase II enzymes. Biochem. J. 2008, 413, 151–161. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carlson, B.A.; Yoo, M.-H.; Tsuji, P.A.; Gladyshev, V.N.; Hatfield, D.L. Mouse Models Targeting Selenocysteine tRNA Expression for Elucidating the Role of Selenoproteins in Health and Development. Molecules 2009, 14, 3509-3527. https://doi.org/10.3390/molecules14093509
Carlson BA, Yoo M-H, Tsuji PA, Gladyshev VN, Hatfield DL. Mouse Models Targeting Selenocysteine tRNA Expression for Elucidating the Role of Selenoproteins in Health and Development. Molecules. 2009; 14(9):3509-3527. https://doi.org/10.3390/molecules14093509
Chicago/Turabian StyleCarlson, Bradley A., Min-Hyuk Yoo, Petra A. Tsuji, Vadim N. Gladyshev, and Dolph L. Hatfield. 2009. "Mouse Models Targeting Selenocysteine tRNA Expression for Elucidating the Role of Selenoproteins in Health and Development" Molecules 14, no. 9: 3509-3527. https://doi.org/10.3390/molecules14093509
APA StyleCarlson, B. A., Yoo, M.-H., Tsuji, P. A., Gladyshev, V. N., & Hatfield, D. L. (2009). Mouse Models Targeting Selenocysteine tRNA Expression for Elucidating the Role of Selenoproteins in Health and Development. Molecules, 14(9), 3509-3527. https://doi.org/10.3390/molecules14093509