Microwave-Assisted Synthesis and Crystal Structure of Oxo(diperoxo)(4,4'-di-tert-butyl-2,2'-bipyridine)-molybdenum(VI)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and spectroscopic characterisation
2.2. X-ray crystal structure


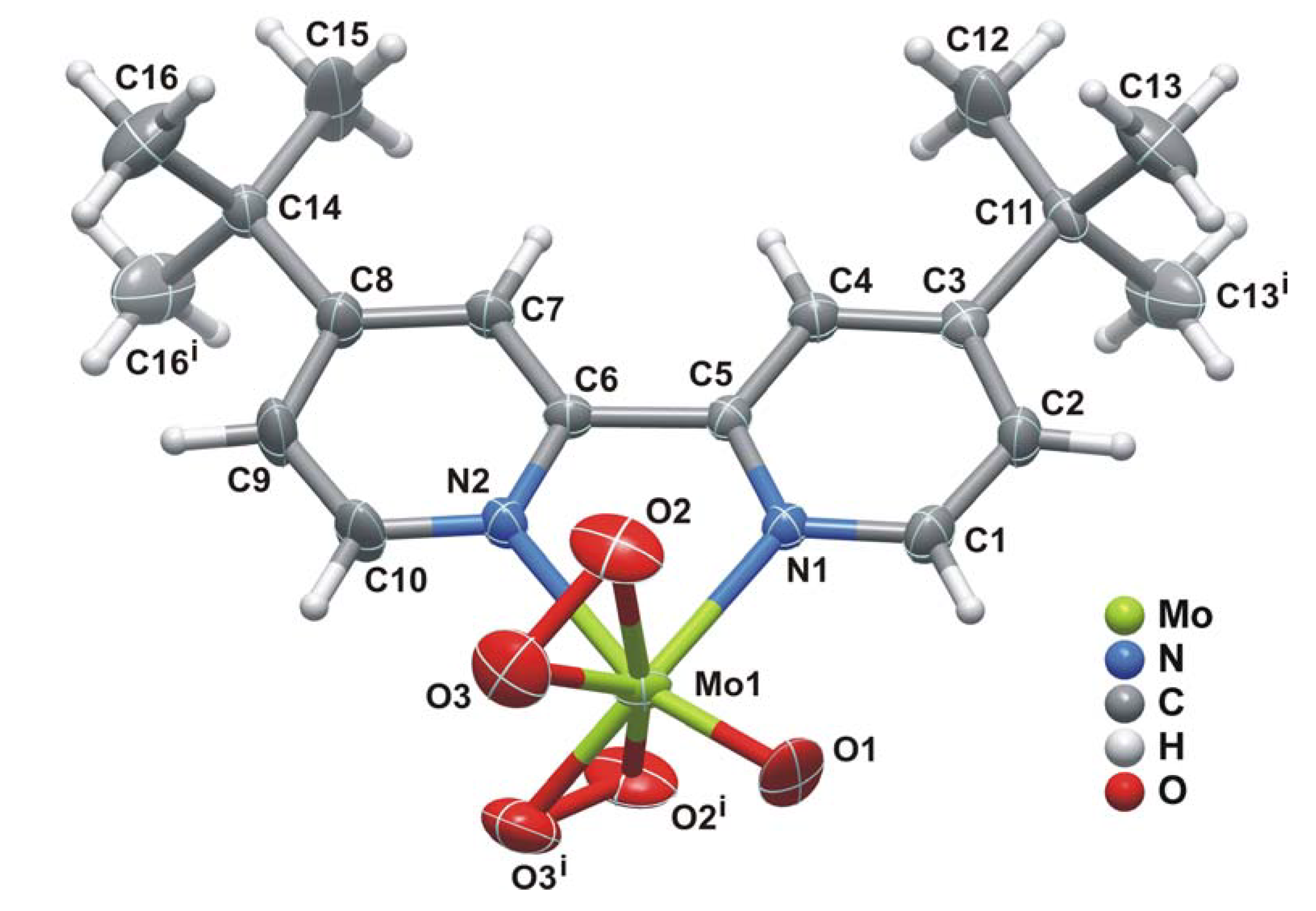{kind=link}
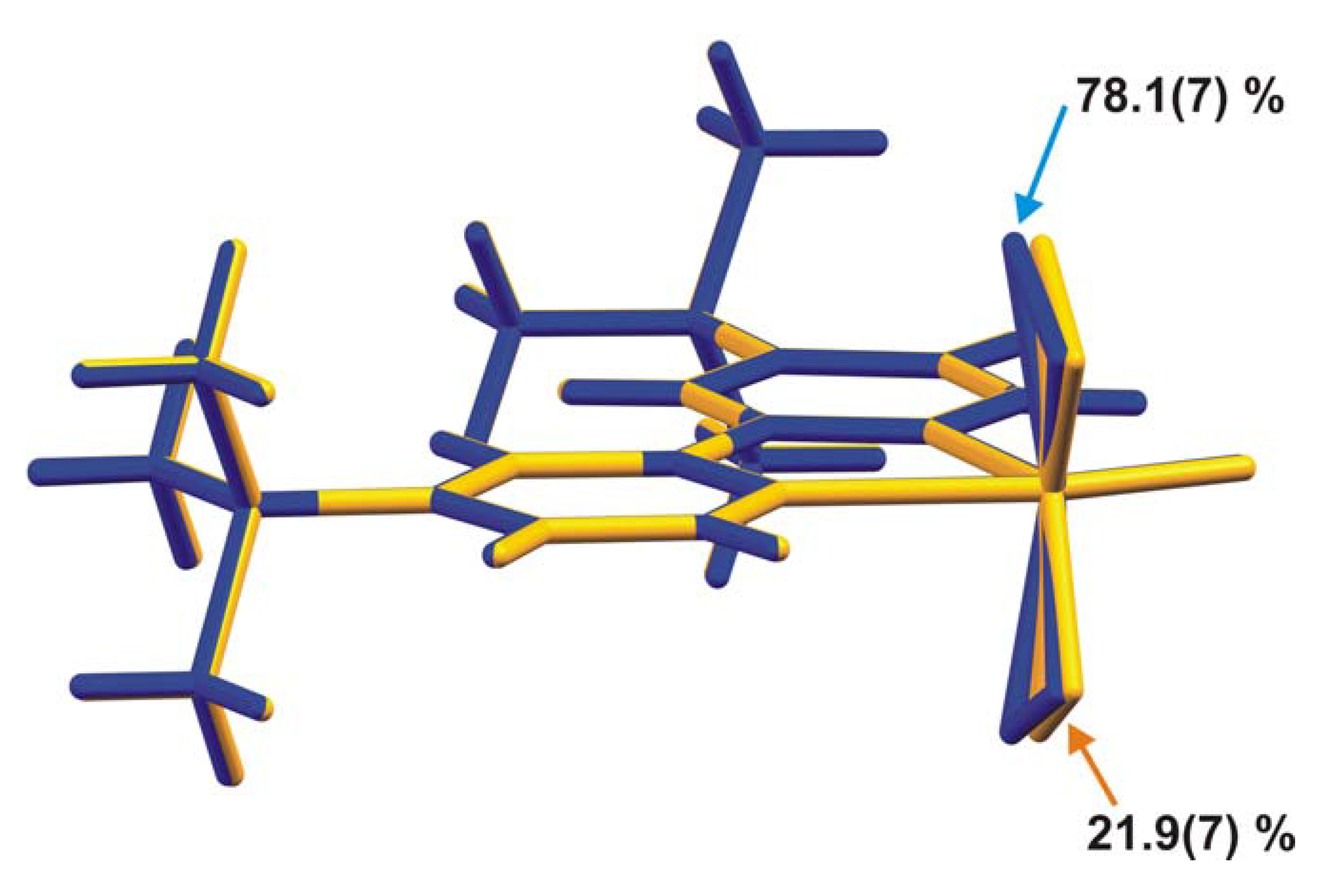{kind=link}
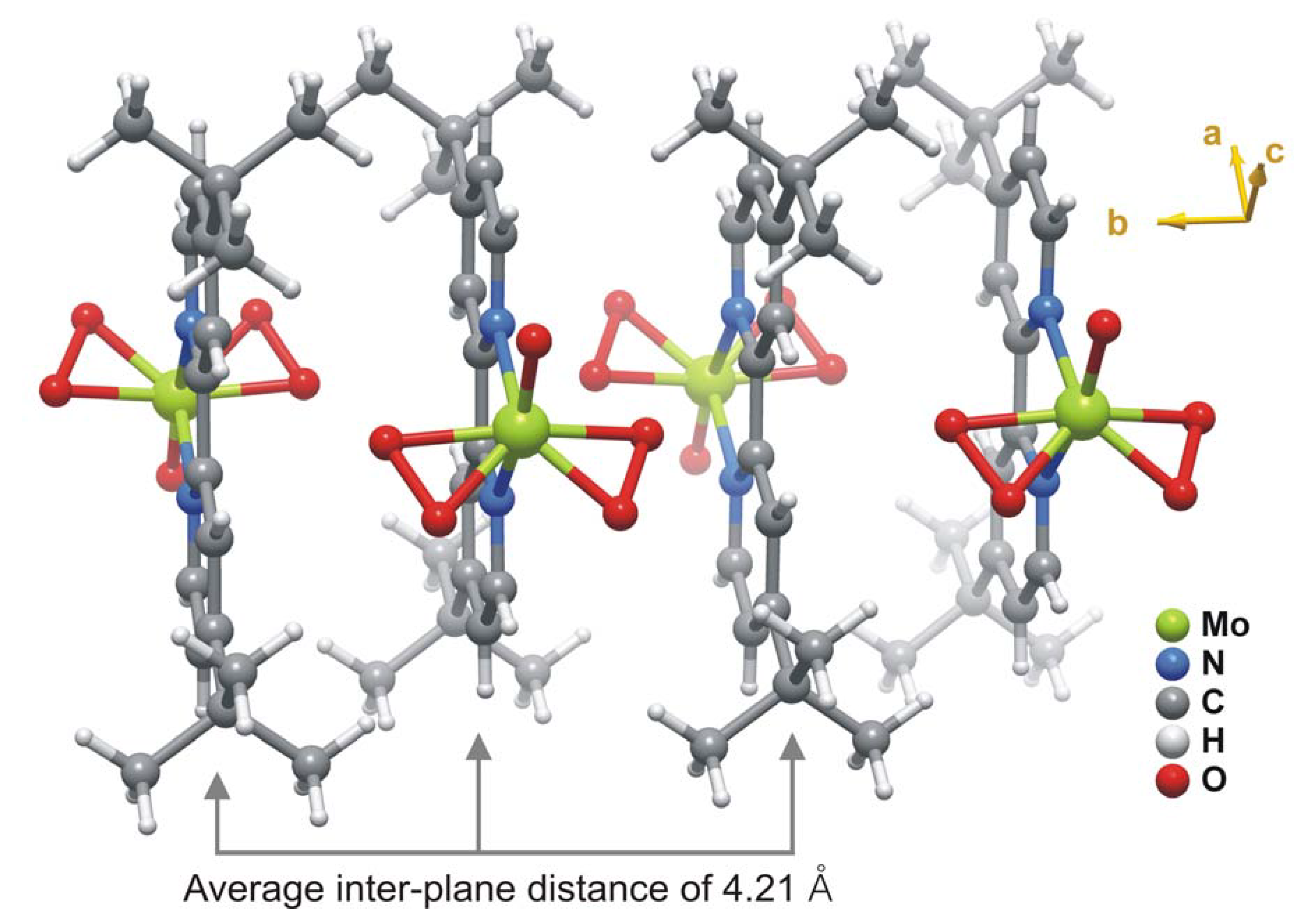{kind=link}
{kind=link}
| Mo1-O1 | 1.698(5) | Mo1-O3' | 1.854(14) |
| Mo1-O1' | 1.757(17) | Mo1-O3 | 1.876(4) |
| Mo1-O2 | 1.939(4) | Mo1-N1 | 2.235(4) |
| Mo1-O2' | 1.89(5) | Mo1-N2 | 2.289(4) |
| O1-Mo1-O2 | 99.10(18) | O1-Mo1-N1 | 89.2(2) |
| O1'-Mo1-O2' | 92.3(17) | O2-Mo1-N1 | 86.20(16) |
| O1-Mo1-O3 | 107.89(18) | O1'-Mo1-N1 | 159.4(6) |
| O1'-Mo1-O3' | 103.8(6) | O2'-Mo1-N1 | 87.4(18) |
| O3-Mo1-O2 | 45.90(19) | O3-Mo1-N1 | 130.31(14) |
| O3'-Mo1-O2' | 46.4(8) | O3'-Mo1-N1 | 90.9(5) |
| O3-Mo1-O2i | 133.4(2) | O1-Mo1-N2 | 159.6(2) |
| O2i -Mo1-O2 | 160.2(3) | O1'-Mo1-N2 | 89.0(6) |
| O2'-Mo1-O2' i | 175(3) | O2-Mo1-N2 | 80.09(17) |
| O3'-Mo1-O2' i | 134.7(12) | O2'-Mo1-N2 | 88.7(10) |
| O3'-Mo1-O3' i | 88.4(10) | O3-Mo1-N2 | 86.34(14) |
| O3i -Mo1-O3 | 89.1(3) | O3'-Mo1-N2 | 132.9(5) |
| O3i -Mo1-O2' | 136.7(17) | N1-Mo1-N2 | 70.39(14) |



3. Experimental
3.1. General
3.2. MoO(O2)2(tbbpy) (1)
3.3. Single crystal X-ray diffraction
| Formula | C18H24MoN2O5 |
| Formula weight | 444.33 |
| Crystal system | Orthorhombic |
| Space group | Pnma |
| a/Å | 12.0720(5) |
| b/Å | 8.4233(3) |
| c/Å | 18.5275(7) |
| Volume/Å3 | 1883.99(13) |
| Z | 4 |
| Dc/g cm−3 | 1.567 |
| μ(Mo-Kα)/mm−1 | 0.727 |
| F(000) | 912 |
| Crystal size/mm | 0.16 × 0.06 × 0.05 |
| Crystal type | Yellow needles |
| θ range | 3.55 to 29.13 |
| Index ranges | −16 ≤ h≤ 11, −11 ≤ k≤10, −25 ≤ l ≤ 25 |
| Data completeness to θ = 29.13 | 98.9% |
| Reflections collected | 24626 |
| Independent reflections | 2676 (Rint = 0.0351) |
| Final R indices [I > 2σ(I)]a,b | R1 = 0.0470, wR2 = 0.1009 |
| Final R indices (all data)a,b | R1 = 0.0612, wR2 = 0.1050 |
| Weighting schemec | m = 0.0200, n = 5.4626 |
| Largest diff. peak and hole | 0.984 and −1.715 eÅ−3 |
 ; b
; b  c
c 
4. Conclusions
Acknowledgements
- Samples Availability: Small quantities of compound 1 are available from the authors on request.
References
- Dickman, M.H.; Pope, M.T. Peroxo and superoxo complexes of chromium, molybdenum, and tungsten. Chem. Rev. 1994, 94, 569–584. [Google Scholar]
- Lane, B.S.; Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2473. [Google Scholar] [CrossRef]
- Butler, A.; Clague, M.J.; Meister, G.E. Vanadium peroxide complexes. Chem. Rev. 1994, 94, 625–638. [Google Scholar]
- Bortolini, O.; Conte, V. Vanadium (V) peroxocomplexes: Structure, chemistry and biological implications. J. Inorg. Biochem. 2006, 99, 1549–1557. [Google Scholar] [CrossRef]
- Bayot, D.; Devillers, M. Peroxo complexes of niobium(V) and tantalum(V). Coord. Chem. Rev. 2006, 250, 2610–2626. [Google Scholar] [CrossRef]
- Kühn, F.E.; Herrmann, W.A. Rhenium-oxo and rhenium-peroxo complexes in catalytic oxidations. In Structure and Bonding; Meunier, B., Ed.; Springer-Verlag: Heidelberg, Berlin, Germany, 2000; Volume 97, pp. 213–236. [Google Scholar]
- Deubel, D.V.; Frenking, G.; Gisdakis, P.; Herrmann, W.A.; Rösch, N.; Sundermeyer, J. Olefin Epoxidation with Inorganic Peroxides. Solutions to four long-standing controversies on the mechanism of oxygen transfer. Acc. Chem. Res. 2004, 37, 645–652. [Google Scholar]
- Burke, A.J. Chiral oxoperoxomolybdenum(VI) complexes for enantioselective olefin epoxidation: Some mechanistic and stereochemical reflections. Coord. Chem. Rev. 2008, 252, 170–175. [Google Scholar]
- Thiel, W.R.; Eppinger, J. Molybdenum-catalyzed olefin epoxidation: Ligand effects. Chem. Eur. J. 1997, 3, 696–705. [Google Scholar] [CrossRef]
- Schlemper, E.O.; Schrauzer, G.N.; Hughes, L.A. Crystal structure of oxo(diperoxo)-bipyridylmolybdenum(VI). Polyhedron 1984, 3, 377–380. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Thiel, W.R.; Kuchler, J.G. Wasserlösliche metallkomplexe und katalysatoren, IV. 2,2-Bipyridin-5-sulfonsäure: Synthese, reinigung, derivate und metallkomplexe. Chem. Ber. 1990, 123, 1953–1961. [Google Scholar]
- Herrmann, W.A.; Thiel, W.R.; Kuchler, J.G.; Behm, J.; Herdtweck, E. Wasserlösliche metallkomplexe und katalysatoren, V. 2,2-Bipyridin-5-sulfonsäure und metallorganische Komplexe: Spektroskopie und strukturchemie. Chem. Ber. 1990, 123, 1963–1970. [Google Scholar]
- Thiel, W.R.; Angstl, M.; Priermeier, T. Substituierte N,N-chelat-liganden-anwendungen in der molybdän-katalysierten olefin-epoxidation. Chem. Ber. 1994, 127, 2373–2379. [Google Scholar] [CrossRef]
- Thiel, W.R.; Priermeier, T. The First olefin-substituted peroxomolybdenum complex: Insight into a new mechanism for the molybdenum-catalyzed epoxidation of olefins. Angew. Chem. Int. Ed. Engl. 1995, 34, 1737–1738. [Google Scholar] [CrossRef]
- Glas, H.; Spiegler, M.; Thiel, W.R. Spectroscopic evidence for intramolecular MoVI(O2)·HO-C hydrogen bonding in solution. Eur. J. Inorg. Chem. 1998, 275–281. [Google Scholar]
- Carreiro, E.P.; Yong-En, G.; Burke, A.J. Synthesis, characterisation and reactivity of oxodiperoxo-[2-(1-pyrazolyl)-6-menthylpyridine]molybdenum(VI): The first chiral 2-(1-pyrazole)pyridineoxodiperoxomolybdenum(VI) Complex. Inorg. Chim. Acta 2006, 359, 1519–1523. [Google Scholar] [CrossRef]
- Brito, J.A.; Gómez, M.; Muller, G.; Teruel, H.; Clinet, J.C.; Duñach, E.; Maestro, M.A. Structural studies of mono- and dimetallic MoVI complexes-A new mechanistic contribution in catalytic olefin epoxidation provided by oxazoline ligands. Eur. J. Inorg. Chem. 2004, 4278–4285. [Google Scholar]
- Maurya, M.R.; Kumar, M.; Sikarwar, S. Polymer-anchored oxoperoxo complexes of vanadium(V), molybdenum(VI) and tungsten(VI) as catalyst for the oxidation of phenol and styrene using hydrogen peroxide as oxidant. React. Funct. Polym. 2006, 66, 808–818. [Google Scholar] [CrossRef]
- Coelho, A.C.; Paz, F.A.A.; Klinowski, J.; Pillinger, M.; Gonçalves, I.S. Microwave assisted synthesis of molybdenum and tungsten tetracarbonyl complexes with a pyrazolylpyridine ligand. Crystal structure of cis-[Mo(CO)4{ethyl[3-(2-pyridyl)-1-pyrazolyl]acetate}]. Molecules 2006, 11, 940–952. [Google Scholar]
- Ardon, M.; Hayes, P.D.; Hogarth, G. Microwave-assisted reflux in organometallic chemistry: Synthesis and structural determination of molybdenum carbonyl complexes. J. Chem. Educ. 2002, 79, 1249–1251. [Google Scholar] [CrossRef]
- VanAtta, S.L.; Duclos, B.A.; Green, D.B. Microwave-assisted synthesis of Group 6 (Cr, Mo, W) zerovalent organometallic carbonyl compounds. Organometallics 2000, 19, 2397–2399. [Google Scholar] [CrossRef]
- Ardon, M.; Hogarth, G.; Oscroft, D.T.W. Organometallic chemistry in a conventional microwave oven: the facile synthesis of group 6 carbonyl complexes. J. Organomet. Chem. 2004, 689, 2429–2435. [Google Scholar] [CrossRef]
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis-a review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Arzoumanian, H.; Agrifoglio, G.; Krentzien, H. Reaction of a tetrathiocyanato-dioxomolybdate(VI) anion with bipyridines. Synthesis of a highly efficient oxo-transfer catalyst. New J. Chem. 1996, 20, 699–705. [Google Scholar]
- Monteiro, B.; Gago, S.; Neves, P.; Valente, A.A.; Gonçalves, I.S.; Pereira, C.C.L.; Silva, C.M.; Pillinger, M. Effect of an ionic liquid on the catalytic performance of thiocyanatodioxomolybdenum(vi) complexes for the oxidation of cyclooctene and benzyl alcohol. Catal. Lett. 2009, 129, 350–357, and references cited therein. [Google Scholar]
- Hinner, M.J.; Grosche, M.; Herdtweck, E.; Thiel, W.R. A Merrifield resin functionalized with molybdenum peroxo complexes: Synthesis and catalytic properties. Z. Anorg. Allg. Chem. 2003, 629, 2251–2257. [Google Scholar] [CrossRef]
- Gago, S.; Rodríguez-Borges, J.E.; Teixeira, C.; Santos, A.M.; J., Zhao; Pillinger, M.; Nunes, C.D.; Petrovski, Ž.; Santos, T.M.; Kühn, F.E.; Romão, C.C.; Gonçalves, I.S. Synthesis, characterization and catalytic studies of bis(chloro)dioxomolybdenum(VI)-chiral diimine complexes. J. Mol. Catal. A Chem. 2005, 236, 1–6. [Google Scholar]
- Kottke, T.; Stalke, D. Crystal handling at low temperatures. J. Appl. Crystallogr. 1993, 26, 615–619. [Google Scholar] [CrossRef]
- APEX2, Data Collection Software Version 2.1-RC13, 2006.
- Cryopad, Remote monitoring and control Version 1.451, 2006.
- SAINT+, Data Integration Engine Version 7.23a, © 1997–2005.
- Sheldrick, G.M. SADABS, Version 2.01,Bruker/Siemens Area Detector Absorption Correction Program; Bruker AXS: Madison, WI, USA, 1998. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS–97, Program for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL–97,Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Amarante, T.R.; Almeida Paz, F.A.; Gago, S.; Gonçalves, I.S.; Pillinger, M.; Rodrigues, A.E.; Abrantes, M. Microwave-Assisted Synthesis and Crystal Structure of Oxo(diperoxo)(4,4'-di-tert-butyl-2,2'-bipyridine)-molybdenum(VI). Molecules 2009, 14, 3610-3620. https://doi.org/10.3390/molecules14093610
Amarante TR, Almeida Paz FA, Gago S, Gonçalves IS, Pillinger M, Rodrigues AE, Abrantes M. Microwave-Assisted Synthesis and Crystal Structure of Oxo(diperoxo)(4,4'-di-tert-butyl-2,2'-bipyridine)-molybdenum(VI). Molecules. 2009; 14(9):3610-3620. https://doi.org/10.3390/molecules14093610
Chicago/Turabian StyleAmarante, Tatiana R., Filipe A. Almeida Paz, Sandra Gago, Isabel S. Gonçalves, Martyn Pillinger, Alírio E. Rodrigues, and Marta Abrantes. 2009. "Microwave-Assisted Synthesis and Crystal Structure of Oxo(diperoxo)(4,4'-di-tert-butyl-2,2'-bipyridine)-molybdenum(VI)" Molecules 14, no. 9: 3610-3620. https://doi.org/10.3390/molecules14093610
APA StyleAmarante, T. R., Almeida Paz, F. A., Gago, S., Gonçalves, I. S., Pillinger, M., Rodrigues, A. E., & Abrantes, M. (2009). Microwave-Assisted Synthesis and Crystal Structure of Oxo(diperoxo)(4,4'-di-tert-butyl-2,2'-bipyridine)-molybdenum(VI). Molecules, 14(9), 3610-3620. https://doi.org/10.3390/molecules14093610