Catalytic Asymmetric Synthesis of Both Enantiomers of 4?Substituted 1,4-Dihydropyridines with the Use of Bifunctional Thiourea-Ammonium Salts Bearing Different Counterions

Abstract
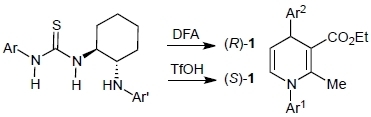
:
1. Introduction

2. Results and Discussion

2.1. Synthesis of chiral bifunctional thioureas 1a-h for Brønsted acid-thiourea co-catalysts

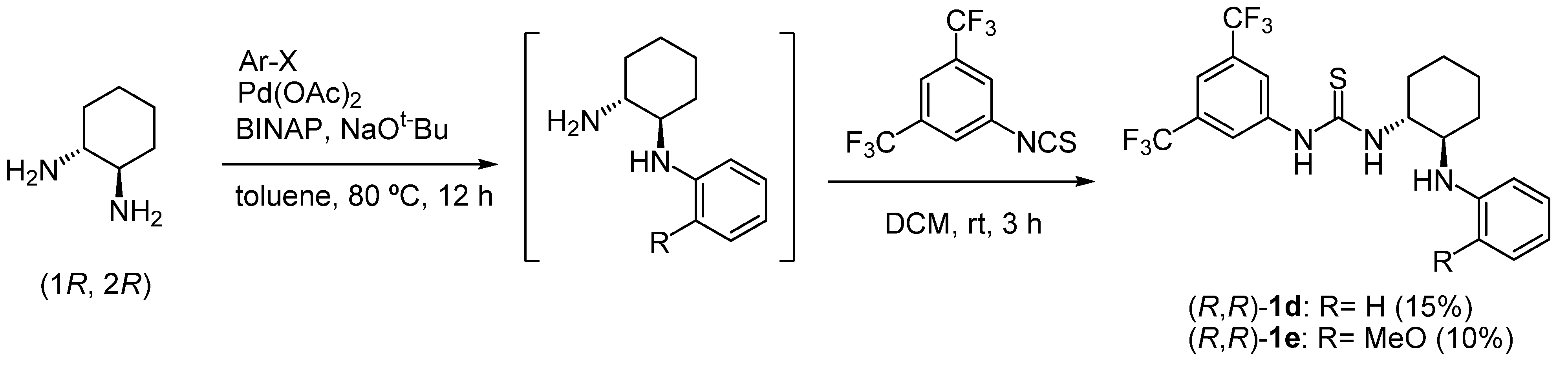

2.2. Brønsted acid-bifunctional thiourea co-catalysts for the synthesis of 3,4-disubstituted 1,4-DHPs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
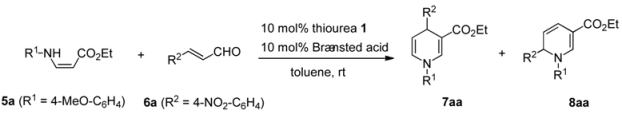 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Thiourea | BA | Time (h) | Conversion (%) b | 7aa | 8aa | |||
| 7aa | 8aa | Yield (%) c | Ee (%) d | Yield (%) c | Ee (%) d | ||||
| 1 | None | HBF4 | 24 | 40 | 6 | - | - | - | |
| 2 | None | TfOH | 24 | 40 | 1 | - | - | - | - |
| 3 | None | TFA | 24 | 64 | 35 | - | - | - | - |
| 4 | None | DFA | 24 | 35 | 64 | - | - | - | - |
| 5 | None | AcOH | 24 | 0 | 0 | - | - | - | - |
| 6 | (R, R)-1a | None | 48 | 0 | 0 | - | - | - | - |
| 7 | (R, R)-1a | DFA | 48 | 0 | 0 | - | - | - | - |
| 8 | (R, R)-1b | DFA | 36 | 46 | 47 | 33 | 1 | 41 | 1 |
| 9 | (R, R)-1c | DFA | 36 | 29 | 56 | 24 | 1 | 48 | 1 |
| 10 | (S, S)-1f | DFA | 24 | 79 | 19 | 72 | 39 (R) | 17 | 0 |
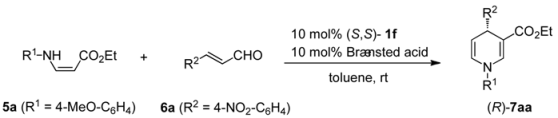 | ||||
|---|---|---|---|---|
| Entry | Brøsted acid | Time (h) | Yield (%) b | Ee (%) c |
| 1 | HBF4 | 48 | 68 | 16 |
| 2 | TfOH | 48 | 78 | 19 |
| 3 | TFA | 46 | 64 | 29 |
| 4 | TCA | 46 | 83 | 34 |
| 5 | DFA | 48 | 72 | 39 |
| 6 | C6F5CO2H | 24 | 61 | 37 |
| 7 | AcOH | 48 | 11 | 78 |
| 8 | BzOH | 48 | 17 | 75 |
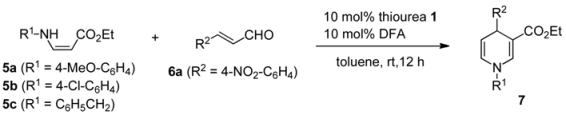 | |||||
|---|---|---|---|---|---|
| Entry | Thiourea 1 | β-Enamino ester 5 | Product 7 | Yield (%) b | Ee (%) c |
| 1 | (S, S)-1f | 5a | 7aa | 86 | 50 (R) |
| 2 | (R, R)-1d | 5a | 7aa | 86 | 42 (S) |
| 3 | (R, R)-1e | 5a | 7aa | 47 | 41 (S) |
| 4 | (S, S)-1g | 5a | 7aa | 91 | 55 (R) |
| 5 | (S, S)-1h | 5a | 7aa | 92 | 50 (R) |
| 6 | (S, S)-1h | 5b | 7ba | 78 | 49 (R) |
| 7 | (S, S)-1h | 5c | 7ca | 83 | 45 (R) |
2.3. Application of new thiourea-ammonium salts to the synthesis of 2,3,4-trisubstituted 1,4-DHP's
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Thiourea | 5 | R1 | R3 | R4 | 6 | R2 | 7 | Yield | Ee |
| (%) | (%) | |||||||||
| 1 | (S, S)-1f | 5d | 4-MeO-C6H4 | Me | OEt | 6a | 4-NO2-C6H4 | 7da | 84 | 61 |
| 2 | (S, S)-1g | 5d | 4-MeO-C6H4 | Me | OEt | 6a | 4-NO2-C6H4 | 7da | 65 | 56 |
| 3 | (S, S)-1h | 5d | 4-MeO-C6H4 | Me | OEt | 6a | 4-NO2-C6H4 | 7da | 93 | 66 |
| 4 | (S, S)-1h | 5e | 4-MeO-C6H4 | Me | Ot-Bu | 6a | 4-NO2-C6H4 | 7ea | 81 | 51 |
| 5 | (S, S)-1h | 5f | 4-MeO-C6H4 | Ph | OEt | 6a | 4-NO2-C6H4 | 7fa | 85 | 61 |
| 6 | (S, S)-1h | 5g | 3,4-MeO-C6H3 | Me | OEt | 6a | 4-NO2-C6H4 | 7ga | 96 | 66 |
| 7 | (S, S)-1h | 5g | 3,4-MeO-C6H3 | Me | OEt | 6b | C6H5 | 7gb | 61 | 44 |
| 8 | (S, S)-1h | 5g | 3,4-MeO-C6H3 | Me | OEt | 6c | 4-MeO-C6H4 | 7gc | 56 | 38 |
| 9 | (S, S)-1h | 5g | 3,4-MeO-C6H3 | Me | OEt | 6d | 4-F-C6H4 | 7gd | 62 | 53 |
| 10 | (S, S)-1h | 5g | 3,4-MeO-C6H3 | Me | OEt | 6e | 3-F-C6H4 | 7ge | 55 | 58 |
| 11 | (S, S)-1h | 5g | 3,4-MeO-C6H3 | Me | OEt | 6f | 2-F-C6H4 | 7gf | 70 | 44 |
| 12 | (S, S)-1h | 5h | 4-Cl-C6H4 | Me | OEt | 6a | 4-NO2-C6H4 | 7ha | 78 | 38 |
| 13 | (S, S)-1h | 5i | C6H4CH2 | Me | OEt | 6a | 4-NO2-C6H4 | 7ia | 81 | 80 |
| 14 | (S, S)-1h | 5j | 4-MeO C6H3CH2 | Me | OEt | 6a | 4-NO2-C6H4 | 7ja | 65 | 77 |
2.4. Utility of thiourea-ammonium salts derived from strong Brønsted acids and anilinothioureas
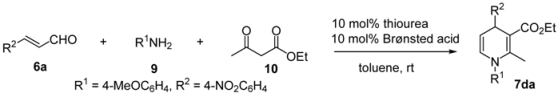 | |||||
|---|---|---|---|---|---|
| Entry | Thiourea | Brønsted acid | Time | Yield (%) b | Ee (%) c |
| 1 | (S, S)-1h | DFA | 48 | 64 | 50 (R) |
| 2 | (S, S)-1h | TFA | 24 | 73 | 49 (R) |
| 3 | (S, S)-1h | TfOH | 60 | 82 | 20 (S) |
| 4 | (S, S)-1h | HBF4 | 72 | 69 | 28 (S) |
| 5 | (S, S)-1f | DFA | 48 | 53 | 43 (R) |
| 6 | (S, S)-1f | TFA | 24 | 76 | 25 (R) |
| 7 | (S, S)-1f | TfOH | 48 | 82 | 50 (S) |
| 8 | (S, S)-1f | HBF4 | 72 | 61 | 37 (S) |
| 9 | (S, S)-1g | DFA | 48 | 49 | 39 (R) |
| 10 | (S, S)-1g | TFA | 24 | 82 | 1 (R) |
| 11 | (S, S)-1g | TfOH | 60 | 77 | 33 (S) |
| 12 | (R, R)-1d | TfOH | 96 | 56 | 39 (R) |
| 13 | (R, R)-1e | TfOH | 96 | 69 | 61 (R) |
| 14 | (R, R)-1e | HBF4 | 72 | 52 | 69 (R) |
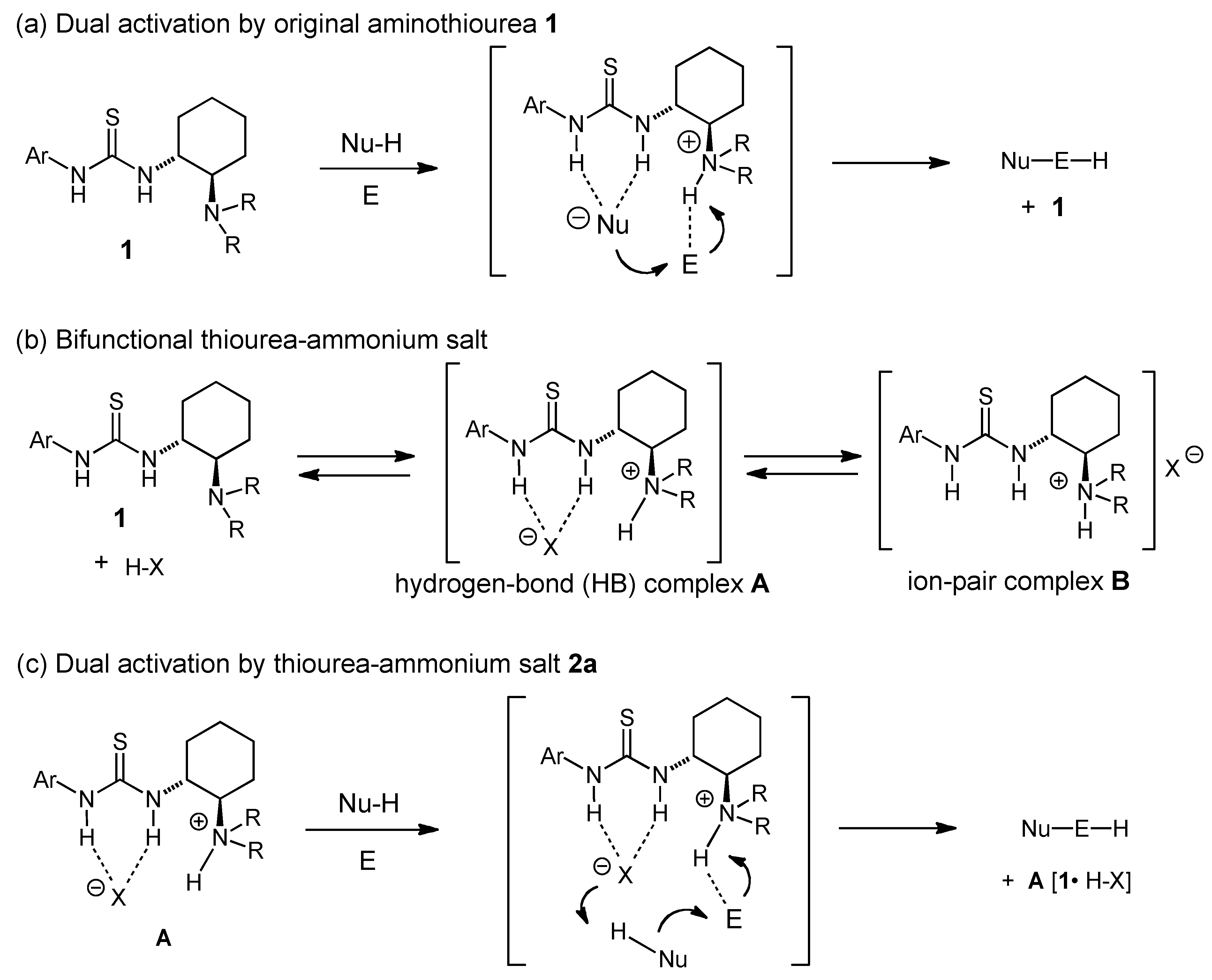
2.5. Proposed reaction mechanism of Brønsted acid-anilinothiourea co-catalysis

3. Experimental
3.1. General
3.2. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-((1R,2R)-2-(2-methoxyphenylamino)cyclohexyl)thiourea [(R, R)-1e]
3.3. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-((1R,2R)-2-(phenylamino)cyclohexyl)thiourea [(R, R)-1d]
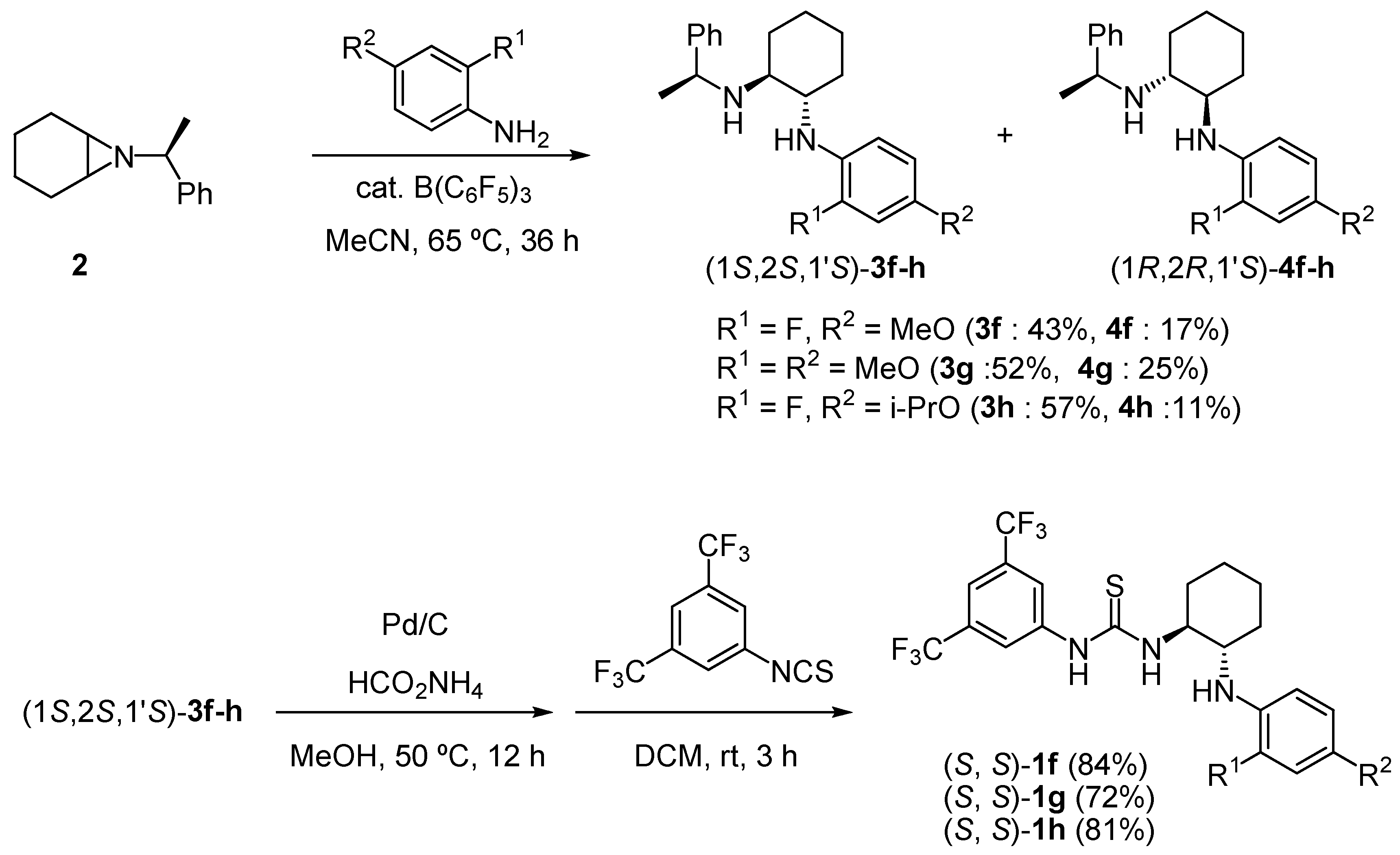
3.4. Synthesis of (1S,2S,1’S)-3f and (1R,2R,1’S)-4f
3.5. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-((1S,2S)-2-(2-fluoro-4-methoxyphenylamino)cyclohexyl)thiourea [(S, S)-1f]
3.6. Synthesis of (1S,2S,1’S)-3g and (1R,2R,1’S)-4g
3.7. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-((1S,2S)-2-(2,4-dimethoxyphenylamino)cyclo- hexyl)thiourea [(S, S)-1g]
3.8. Synthesis of (1S,2S,1’S)-3h and (1R,2R,1’S)-4h
3.9. Synthesis of 1-(3,5-bis(trifluoromethyl)phenyl)-3-((1S,2S)-2-(2-fluoro-4-isopropoxyphenylamino)cyclohexyl)thiourea [(S, S)-1h]
3.10. Preparation of (Z)-ethyl 3-(4-methoxyphenylamino)acrylate (5a)
3.11. Preparation of enamino esters 5b-k
3.12. General Procedure for the reaction of enaminoester 5a with 4-nitrocinnamaldehyde (6a) catalyzed by thiourea 1 – Brønsted acid (Table 1 and Table 2).
3.13. Typical Procedure for the reaction of enaminoester 5a with 4-nitrocinnamaldehyde 6a catalyzed by thiourea (S, S)-1h – difluoro acid (Table 3 and Table 4)
3.14. General Procedure for the reaction of 4-nitro cinnamaldehyde 6a and 4-methoxyaniline (9) with ethyl acetoacetate (10) catalyzed by thiourea 1—Brønsted acid (Table 5)
4. Conclusions
Acknowledgements
- Sample Availability: Contact the authors.
References and Notes
- Ikariya, T.; Blacker, A.J. Asymmetric Transfer Hydrogenation of Ketones with Bifunctional Transition Metal-Based Molecular Catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef]
- Ikariya, T.; Murata, K.; Noyori, R. Bifunctional trans metal-based molecular catalysts for asymmetric syntheses. Org. Biomol. Chem. 2006, 4, 393–406. [Google Scholar] [CrossRef]
- Shibasaki, M.; Sasai, H.; Arai, T. Asymmetric Catalysis with Heterobimet allic Compounds. Angew. Chem., Int. Ed. 1997, 36, 1236–1256. [Google Scholar] [CrossRef]
- Kitamura, M.; Suga, S.; Kawai, K.; Noyori, R. Catalytic asymmetric induction. Highly enantioselective addition of dialkylzincs to aldehydes. J. Am. Chem. Soc. 1986, 108, 6071–6072. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, L. Asymmetric Acid-Base Bifunctional Catalysis with Organic Molecules. In Catalytic Asymmetric Synthesis, 3rd; Ojima, I., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 59–94. [Google Scholar]
- Taylar, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Hamza, A.; Schubert, G.; Soós, T.; Pápai, I. Theoretical Studies on the Bifunctionality of Chiral Thiourea-Based Organocatalysts: Competing Routes to C−C Bond Formation. J. Am. Chem. Soc. 2006, 128, 13151–13160. [Google Scholar] [CrossRef]
- Zhu, R.; Zhang, D.; Wu, J.; Liu, C. Theoretical study on the enantioselective a-amination reaction of 1,3-dicarbonyl compounds catalyzed by a bifunctional-urea. Tetrahetron Asymmetry 2007, 18, 1655–1662. [Google Scholar] [CrossRef]
- Takemoto, Y. Development of Chiral Thiourea Catalysts and Its Application to Asymmetric Catalytic Reactions. Chem. Pharm. Bull. 2010, 58, 593–601. [Google Scholar] [CrossRef]
- Miyabe, H,; Takemoto, Y. Discovery and Application of Asymmetric Reaction by Multi-Functional Thioureas. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. [Google Scholar] [CrossRef]
- Takemoto, Y. Recognition and activation by ureas and thioureas: Stereoselective reactions using ureas and thioureas as hydrogen-bonding donors. Org. Biomol. Chem. 2005, 3, 4299–4306. [Google Scholar] [CrossRef]
- Yamamoto, H.; Payette, J.N. Brønsted acid, H-Bond Donors, and Combined Acid systems in Asymmetric Catalysis. In Hydrogen Bonding in Organic Synthesis, 1st; Pihko, P.M., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 73–140. [Google Scholar]
- Ishihara, K.; Nakano, K. Design of an Organocatalyst for the Enantioselective Diels−Alder Reaction with α-Acyloxyacroleins. J. Am. Chem. Soc. 2005, 127, 10504–10505. [Google Scholar] [CrossRef]
- Bolm, C.; Rantanen, T.; Schiffers, I.; Zani, L. Protonated Chiral Catalysts: Versatile Tools for Asymmetric Synthesis. Angew. Chem. Int. Ed. 2005, 44, 1758–1763. [Google Scholar] [CrossRef]
- Nakadai, M.; Saito, S.; Yamamoto, H. Diversity-based strategy for discovery of environmentally bengin organocatalyst: Diamine-protonic acid catalysts for asymmetric direct aldol reaction. Tetrahedoron 2002, 58, 8167–8177. [Google Scholar] [CrossRef]
- Yoshida, K.; Inokuma, T.; Tkakasu, K.; Takemoto, Y. Brønsted Acid-Thiourea Co-catalysis: Asymmetric Synthesis of Functionalized 1,4-Dihydropyridines from β-Enamino Esters and α,β-Unsaturated Aldehydes. Synlett 2010, 1865–1869. [Google Scholar]
- Singh, R.P.; Foxman, B.M.; Deng, L. Asymmetric Vinylogous Aldol Reaction of Silyloxy Furans with a Chiral Organic Salt. J. Am. Chem. Soc. 2010, 132, 9558–9560. [Google Scholar] [CrossRef]
- Uyeda, C.; Jacobsen, E.N. Bifunctional Asymmetric Catalysis with Hydrogen Chloride: Enantioselective Ring Opening of Aziridines Catalyzed by a Phosphinothiourea. Synlett 2009, 1680–1684. [Google Scholar]
- Maiti, S.; Menendez, J.C. A Mild Protocol for the Efficient Synthesis of 5,6-Unsubstituted 1,4-Dihydropyridines. Synlett 2009, 2249–2252. [Google Scholar]
- Wan, J.-P.; Gan, S.-F.; Sun, G.-L.; Pan, Y.-J. Novel Regioselectivity: Three-Component Cascade Synthesis of Unsymmetrical 1,4- and 1,2-Dihydropyridines. J. Org. Chem. 2009, 74, 2862–2865. [Google Scholar] [CrossRef]
- Bartoli, G.; Babiuch, K.; Bosco, M.; Carlone, A.; Galzerano, P.; Melchiorre, P.; Sambri, L. Magnesium Perchlorate as Efficient Lewis Acid: A Simple and Convenient Route to 1,4-Dihydropyridines. Synlett 2007, 2897–2901. [Google Scholar]
- Sridharan, V.; Perumal, P.T.; Avendano, C.; Menendez, J.C. A new three-component domino synthesis of 1,4-dihydropyridines. Tetrahedron 2007, 63, 4407–4413. [Google Scholar] [CrossRef]
- Vanden Eynde, J.J.; Mayence, A. Synthesis and Aromatization of Hantzsch 1,4-Dihydropyridines under Microwave Irradiation. An Overview. Molecules 2003, 8, 381–391. [Google Scholar] [CrossRef]
- Jiang, J.; Yu, J.; Sun, X.-X.; Rao, Q.-Q.; Gong, L.-Z. Organocatalytic Asymmetric Three-Component Cyclization of Cinnamaldehydes and Primary Amines with 1,3-Dicarbonyl Compounds: Straightforward Access to Enantiomerically Enriched Dihydropyridines. Angew. Chem. Int. Ed. 2008, 47, 2458–2462. [Google Scholar] [CrossRef]
- Moreau, J.; Duboc, A.; Hubert, C.; Hurvois, J.-P.; Renaud, J.-L. Metal-free Brønsted acids catalyzed synthesis of functional 1,4-dihydropyridines. Tetrahedron Lett. 2007, 48, 8647–8450. [Google Scholar] [CrossRef]
- Kumar, A.; Marurya, R.A. Organocatalysed three-component domino synthesis of 1,4-dihydropyridines under solvent free conditions. Tetrahedron 2008, 64, 3477–3482. [Google Scholar] [CrossRef]
- Parrodi, C.A.; Moreno, G.E.; Quintero, L.; Juaristi, E. Application of phosphorylated reagents derived from N,N′-di-[(S)-α-phenylethyl]-cyclohexane-1,2-diamines in the determination of the enantiomeric purity of chiral alcohols . Tetrahedron Asymmetry 1998, 9, 2093–2099. [Google Scholar] [CrossRef]
- Vachal, P.; Jacobsen, E.N. Structure-Based Analysis and Optimization of a Highly Enantioselective Catalyst for the Strecker Reaction. J. Am. Chem. Soc. 2002, 124, 10012–10014. [Google Scholar] [CrossRef]
- Qian, D.; Weiran, Y.; Xumu, Z. Efficient Rhodium-Catalyzed Asymmetric Hydrogenation for the Synthesis of a New Class of N-Aryl β-Amino Acid Derivatives. Org. Lett. 2005, 7, 5343–5345. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yoshida, K.; Inokuma, T.; Takasu, K.; Takemoto, Y. Catalytic Asymmetric Synthesis of Both Enantiomers of 4?Substituted 1,4-Dihydropyridines with the Use of Bifunctional Thiourea-Ammonium Salts Bearing Different Counterions. Molecules 2010, 15, 8305-8326. https://doi.org/10.3390/molecules15118305
Yoshida K, Inokuma T, Takasu K, Takemoto Y. Catalytic Asymmetric Synthesis of Both Enantiomers of 4?Substituted 1,4-Dihydropyridines with the Use of Bifunctional Thiourea-Ammonium Salts Bearing Different Counterions. Molecules. 2010; 15(11):8305-8326. https://doi.org/10.3390/molecules15118305
Chicago/Turabian StyleYoshida, Kohzo, Tsubasa Inokuma, Kiyosei Takasu, and Yoshiji Takemoto. 2010. "Catalytic Asymmetric Synthesis of Both Enantiomers of 4?Substituted 1,4-Dihydropyridines with the Use of Bifunctional Thiourea-Ammonium Salts Bearing Different Counterions" Molecules 15, no. 11: 8305-8326. https://doi.org/10.3390/molecules15118305
APA StyleYoshida, K., Inokuma, T., Takasu, K., & Takemoto, Y. (2010). Catalytic Asymmetric Synthesis of Both Enantiomers of 4?Substituted 1,4-Dihydropyridines with the Use of Bifunctional Thiourea-Ammonium Salts Bearing Different Counterions. Molecules, 15(11), 8305-8326. https://doi.org/10.3390/molecules15118305