Synthesis of a New Chiral Pyrrolidine †


Abstract
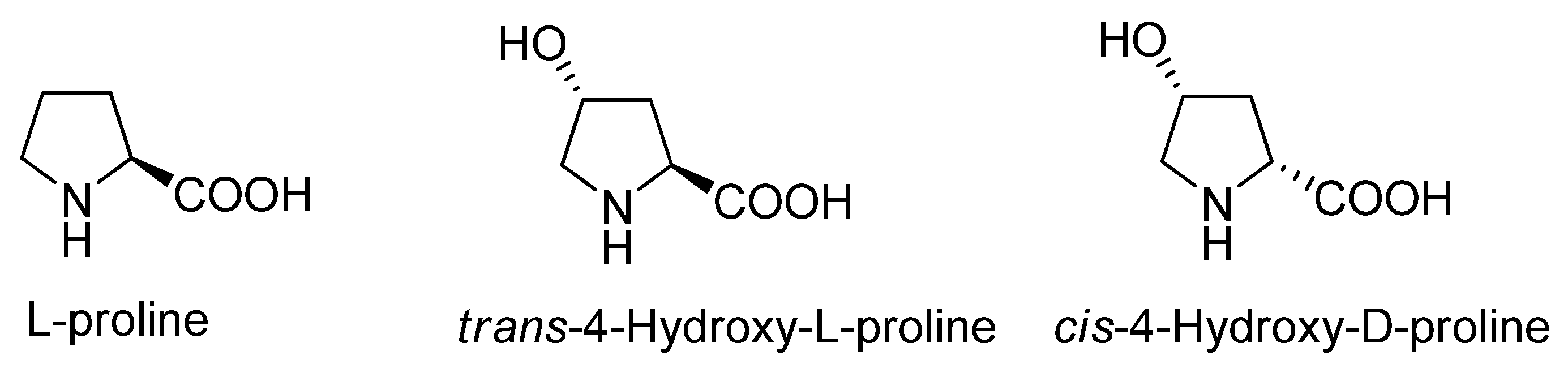
:1. Introduction
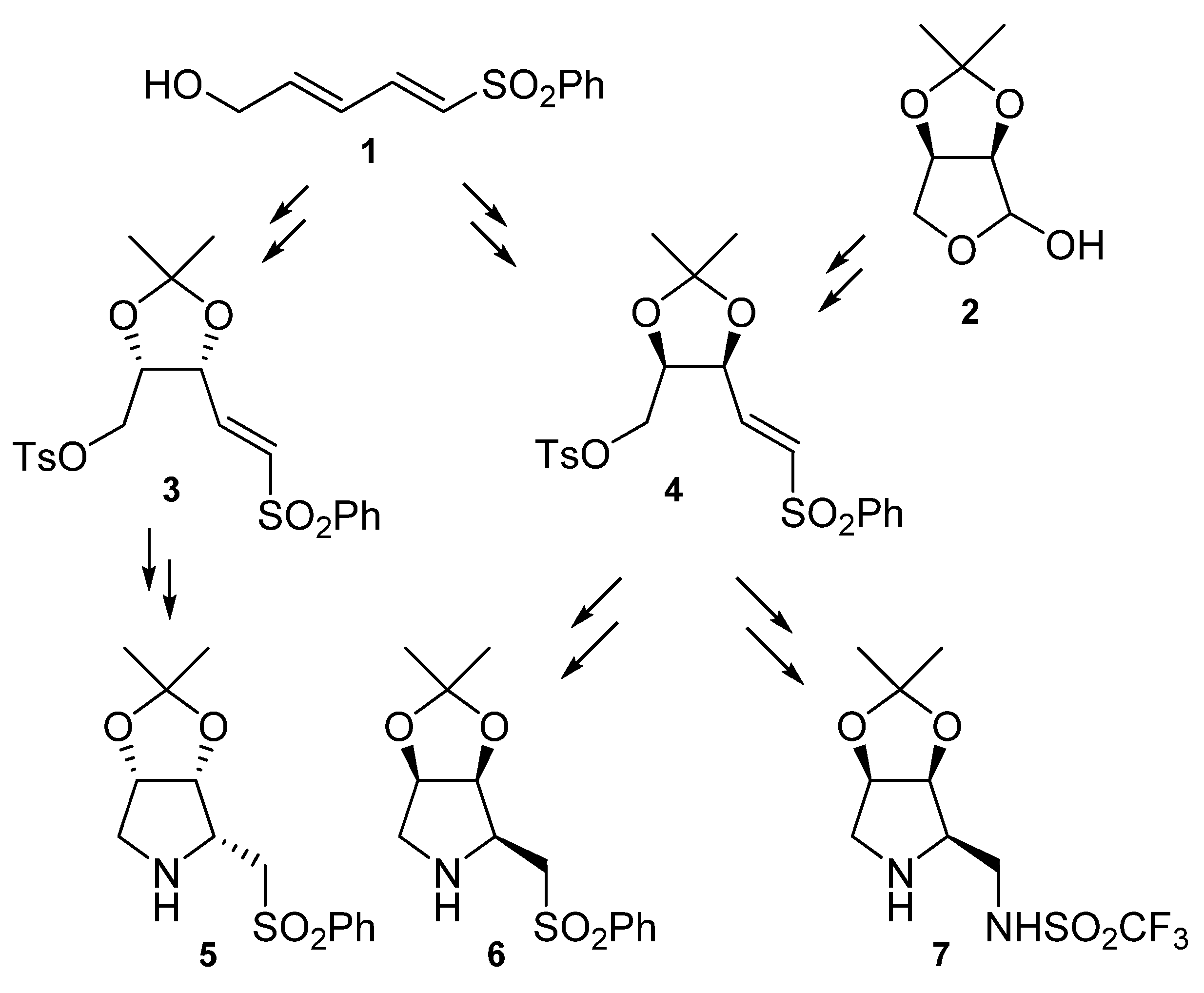
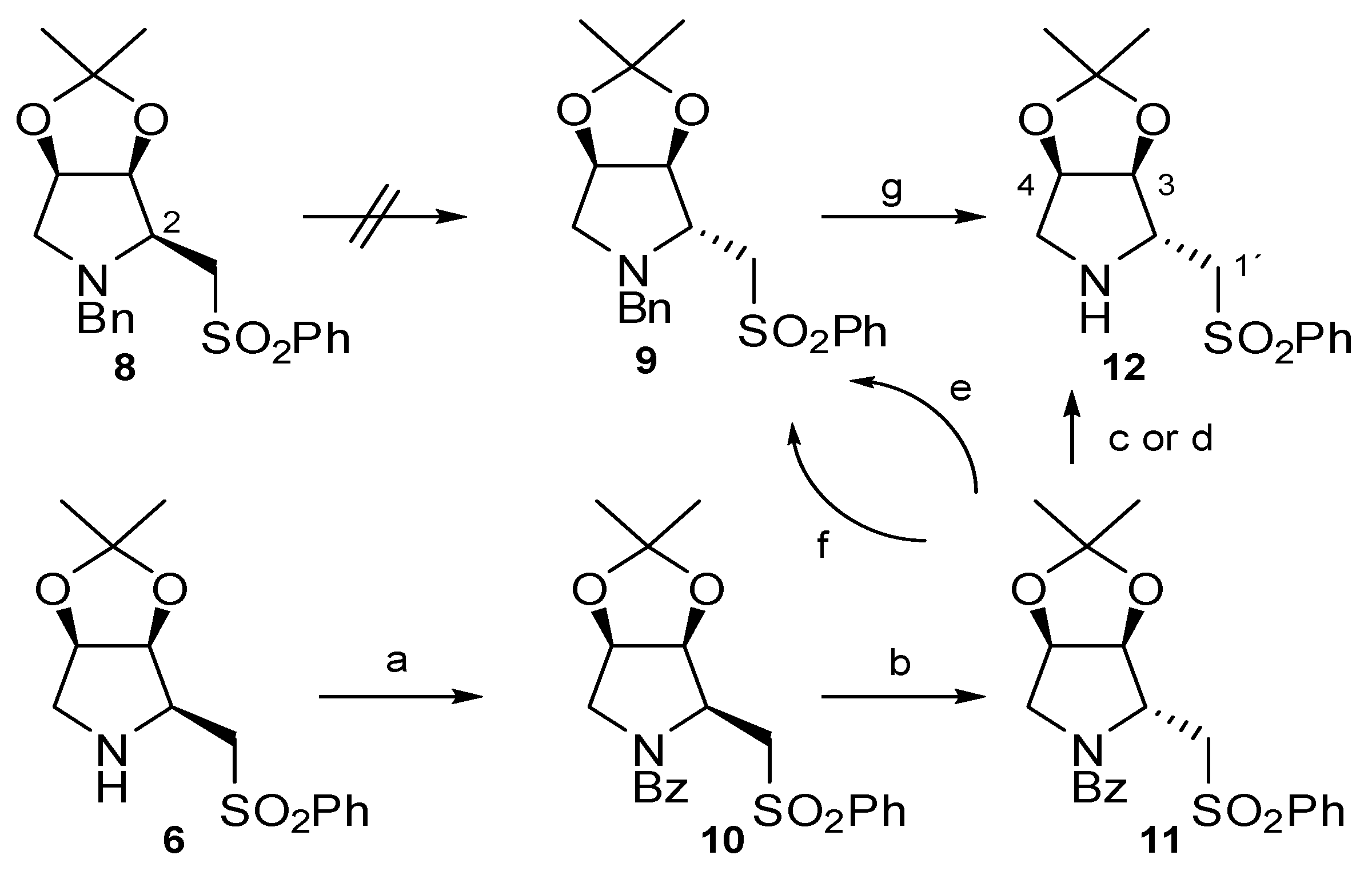
2. Results and Discussion
3. Conclusions
4. Experimental
4.1. General
4.2. Preparation of (2S,3S,4R)-N-Benzoyl-2-phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (10)
4.3. Preparation of (2R,3S,4R)-N-Benzoyl-2-phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (11)
4.4. Preparation of (2R,3S,4R)-N-Benzyl-2-phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (9)
4.5. Preparation of (2R,3S,4R)-2-Phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (12)
4.6. Preparation of (3S,4R)-3,4-Isopropylidenedioxypyrroline-1-oxide (13)
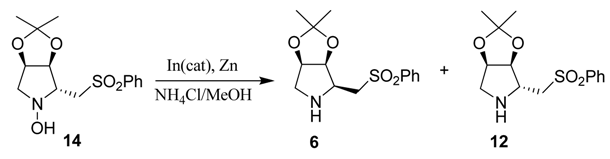
4.7. Preparation of (2R,3S,4R)-2-Phenylsulfonylmethyl-1-hydroxy-3,4-isopropylidenedioxypyrrolidine (14)
4.8. Preparation of (2S,3S,4R)-2-Phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (6)
4.9. Preparation of (2R,3S,4R)-2-Phenylsulfonylmethyl-1-hydroxy-3,4-isopropylidenedioxypyrrolidine (15)
4.10. Preparation of (2R,3S,4R)-2-Phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (12)
4.11. (2S,1´R)-2-[1´-Phenyl-2´-nitroethyl]-cyclohexanone
4.12. (2R,1´S)-2-[1´-Phenyl-2´-nitroethyl]-cyclohexanone
Acknowledgments
References and Notes
- Dalko, P.I.; Moisan, L. Enantioselective Organocatalysis. Angew. Chem. Int. Ed. Engl. 2001, 113, 3726–3748. [Google Scholar] [CrossRef]
- List, B. Asymmetric Aminocatalysis. Synlett 2001, 1675–1686. [Google Scholar] [CrossRef]
- Brown, S.P.; Brochu, M.P.; Sinz, C.J.; MacMillan, D.W.C. The Direct and Enantioselective Organocatalytic -Oxidation of Aldehydes. J. Am. Chem. Soc. 2003, 125, 10808–10809. [Google Scholar] [CrossRef] [PubMed]
- Pidathala, C.; Hoang, L.; Vignola, N.; List, B. Direct Catalytic Asymmetric Enolexo Aldolizations. Angew. Chem. Int. Ed. Engl. 2003, 42, 2785–2788. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis. From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Mase, N.; Barbas, C.F. Design and Use of Fluorogenic Aldehydes for Monitoring the Progress of Aldehyde Transformations. J. Am. Chem. Soc. 2004, 126, 3692–3693. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, Y.; Sasaoka, A.; Shimomoto, A.; Fujioka, S.; Kotsuki, H. High-Pressure-Promoted Asymmetric Aldol Reactions of Ketones with Aldehydes Catalyzed by L-Proline. Synlett 2003, 1655–1658. [Google Scholar]
- Hayashi, Y.; Yamaguchi, J.; Sumiya, T.; Shoji, M. Direct Proline-Catalyzed Asymmetric -Aminoxylation of Ketones. Angew. Chem. Int. Ed. Engl. 2004, 43, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
- Mangion, I.K.; Northrup, A.B.; MacMillan, D.W.C. The Importance of Iminium Geometry Control in Enamine Catalysis: Identification of a New Catalyst Architecture for Aldehyde-Aldehyde Couplings. Angew. Chem. Int. Ed. Engl. 2004, 43, 6722–6724. [Google Scholar] [CrossRef] [PubMed]
- Cobb, A.J.A.; Longbottom, D.A.; Shaw, D.M.; Ley, S.V. 5-Pyrrolidin-2-yltetrazole as an asymmetric organocatalyst for the addition of ketones to nitro-olefins. Chem. Commun. 2004, 1808–1809. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, J.; Li, H. Direct, Highly Enantioselective Pyrrolidine Sulfonamide Catalyzed Michael Addition of Aldehydes to Nitrostyrenes. Angew. Chem. Int. Ed. Engl. 2005, 44, 1369–1371. [Google Scholar] [CrossRef] [PubMed]
- Andrey, O.; Alexakis, A.; Tomassini, A.; Bernardinelli, G. The Use of N-Alkyl-2,2´-bipyrrolidine Derivatives as Organocatalysts for the Asymmetric Michael Addition of Ketones and Aldehydes to Nitroolefins. Adv. Synth. Catal. 2004, 346, 1147–1168. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Diez, D.; Beneitez, M.T.; Marcos, I.S.; Garrido, N.M.; Basabe, P.; Urones, J.G. Enantioselective Synthesis of a 2,3,4-Trisubstituted Pyrrolidine from 1-Hydroxymethyl-4-phenylsulfonylbutadiene. Synlett 2001, 655–657. [Google Scholar] [CrossRef]
- Diez, D.; Beneitez, M.T.; Moro, R.F.; Marcos, I.S.; Basabe, P.; Garrido, N.M.; Urones, J.G. Regio- and stereoselective ring opening of epoxides Enantioselective synthesis of 2,3,4-trisubstituted five-membered heterocycles. Tetrahedron Asymmetry 2002, 13, 639–646. [Google Scholar] [CrossRef]
- Diez, D.; Beneitez, M.T.; Moro, R.F.; Marcos, I.S.; Basabe, P.; Garrido, N.M.; Urones, J.G. Synthesis of Vinylsulfone Derivatives of Sugars: An Easy Preparation of (2R,3S,4E)-5-Benzenesulfonyl-2,3-iso-propylidene-dioxy-pent-4-en-1-yl-tosylate. Synlett 2003, 729–731. [Google Scholar] [CrossRef]
- Diez, D.; Núñez, M.G.; Moro, R.F.; Marcos, I.S.; Basabe, P.; Broughton, H.B.; Urones, J.G. Organocatalytic Synthesis of an Alkyltetrahydropyran. Synlett 2009, 390–394. [Google Scholar] [CrossRef]
- Nising, C.F.; Bräse, S. The oxa-Michael reaction: From recent developments to applications in natural product synthesis. Chem. Soc. Rev. 2008, 37, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Diez, D.; Gil, M.J.; Moro, R.F.; Marcos, I.S.; García, P.; Basabe, P.; Garrido, N.M.; Broughton, H.B.; Urones, J.G. A new class of chiral pyrrolidine for asymmetric Michael addition reactions. New mechanism via simple 4+2 type attack of the enamine on the trans-nitrostyrene. Tetrahedron 2007, 63, 740–747. [Google Scholar] [CrossRef]
- Diez, D.; Gil, M.J.; Moro, R.F.; Garrido, N.M.; Marcos, I.S.; Basabe, P.; Sanz, F.; Broughton, H.B.; Urones, J.G. Chemistry of sulfones: Synthesis of a new chiral nucleophilic catalyst. Tetrahedron Asymmetry 2005, 16, 2980–2985. [Google Scholar] [CrossRef]
- Cicchi, S.; Marradi, M.; Vogel, P.; Goti, A. One-Pot Synthesis of cyclic Nitrones and Their Conversion to Pyrrolizidines: 7a-epi-Crotanecine Inhibits -Mannosidases. J. Org. Chem. 2006, 71, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Cicchi, S.; Corsi, M.; Brandi, A.; Goti, A. Straigtforward Synthesis of Enantiomerically Pure (3S,4R)-and (3R,4S)-3,4-Isopropylidendioxypyrroline 1-Oxide, Precursors of Fucntionalized cis-Dihydroxy Azaheterocycles, by a Novel “One-Pot” Procedure. J. Org. Chem. 2002, 67, 1678–1681. [Google Scholar] [CrossRef]
- Revuelta, J.; Cicchi, S.; Goti, A.; Brandi, A. Enantiopure Cyclic Nitrones: A Useful Class of Building Blocks for Asymmetric Syntheses. Synthesis 2007, 485–504. [Google Scholar] [CrossRef]
- McCaig, A.E.; Meldrum, K.P.; Wightman, R.H. Synthesis of Trihydroxylated Pyrrolizidines using Cycloaddition Reactions of Functionalized Cyclic Nitrones, and the Synthesis of (+)- and (-)-Lentiginosine. Tetrahedron 1998, 54, 9429–9446. [Google Scholar] [CrossRef]
- Hall, A.; Meldrum, K.P.; Therond, P.R.; Wightman, R.H. Synthesis of Hydroxylated Pyrrolizidines Related to Alexine using Cycloaddition Reactions of Functionalized Cyclic Nitrones. Synlett 1997, 123–125. [Google Scholar]
- Closa, M.; Wightman, R.H. Synthesis of (3S,4R)-3,4-Isopropylidenedioxy-1-pyrroline-N-oxide, an Enantiopure Functionalized cyclic nitrone; Cycloaddition reactions with Dimethyl maleate and Dimethyl fumarate. Synth. Commun. 1998, 28, 3443–3450. [Google Scholar] [CrossRef]
- Cardona, F.; Moreno, G.; Guarna, F.; Vogel, P.; Schuetz, C.; Merino, P.; Goti, A. New Concise Total Synthesis of (+)-Lentiginosine and Some Structural Analogues. J. Org. Chem. 2005, 70, 6552–6555. [Google Scholar] [CrossRef] [PubMed]
- Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F.M.; Goti, A. Stereocontrolled Cyclic Nitrone Cycloaddition Strategy for the Synthesis of Pyrrolizidine and Indolizine Alkaloids. Chem. Eur. J. 2009, 15, 7808–7821. [Google Scholar] [CrossRef] [PubMed]
- Cicchi, S.; Bonami, M.; Cardona, F.; Revuelta, J.; Goti, A. Indium-Mediated Reduction of Hydroxylamines to Amines. Org. Lett. 2003, 5, 1773–1776. [Google Scholar] [CrossRef] [PubMed]
- Cobb, A.J.A.; Shaw, D.M.; Longbottom, D.A.; Gold, J.B.; Ley, S.V. Organocatalysis with proline derivatives: Improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions. Org. Biomol. Chem. 2005, 3, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Alza, E.; Cambeiro, X.C.; Jimeno, C.; Pericàs, M.A. Highly Enantioselective Michael Additions in Water Catalyzed by a PS-Supported Pyrrolidine. Org. Lett. 2007, 9, 3717–3720. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 6 and 12 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Zn Equiv. | Ta | t(h) | η (%) | Ratio 6/12 |
|---|---|---|---|---|---|
| 1 | 6 | Reflux | 1 | 15 | 40/60 |
| 2 | 4 | Reflux | 2 | 10 | 36/64 |
| 3 | 4 | Reflux | 1 | 15 | 30/70 |
| 4 | 4 | Reflux | 0.5 | NDa | NDa |
| 5 | 2 | Reflux | 1 | NDa | NDa |
| 6 | 2 | Reflux | 1.5 | 10 | 40/60 |
| 7 | 2 | Reflux | 2 | 20 | 15/85 |
| 8 | 4 | r.t. | 5.5 | 40 | 5/95 |
| 9 | 3 | r.t. | 4 | 8 | 45/55 |
| 10 | 2 | r.t. | 4 | NDa | NDa |
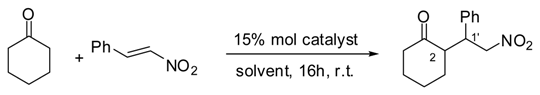
| Entry[a] | Solv. | Catalyst | Yield [%][b] | d.r.[%] [c] | ee[%][d] | Conf. |
|---|---|---|---|---|---|---|
| 1 | CHCl3 | 6 | - | - | - | - |
| 2 | CHCl3 | 12 | 35 | >95 | 38 | 2R, 1’S |
| 3 | DMSO | 6 | 20 | >95 | 69 | 2S, 1’R |
| 4 | DMSO | 12 | - | - | - | - |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Flores, M.F.; Núñez, M.G.; Moro, R.F.; Garrido, N.M.; Marcos, I.S.; Iglesias, E.F.; García, P.; Díez, D. Synthesis of a New Chiral Pyrrolidine. Molecules 2010, 15, 1501-1512. https://doi.org/10.3390/molecules15031501
Flores MF, Núñez MG, Moro RF, Garrido NM, Marcos IS, Iglesias EF, García P, Díez D. Synthesis of a New Chiral Pyrrolidine. Molecules. 2010; 15(3):1501-1512. https://doi.org/10.3390/molecules15031501
Chicago/Turabian StyleFlores, Mari Fe., Marta G. Núñez, Rosalina F. Moro, Narciso M. Garrido, Isidro S. Marcos, Enrique F. Iglesias, Pilar García, and David Díez. 2010. "Synthesis of a New Chiral Pyrrolidine" Molecules 15, no. 3: 1501-1512. https://doi.org/10.3390/molecules15031501
APA StyleFlores, M. F., Núñez, M. G., Moro, R. F., Garrido, N. M., Marcos, I. S., Iglesias, E. F., García, P., & Díez, D. (2010). Synthesis of a New Chiral Pyrrolidine. Molecules, 15(3), 1501-1512. https://doi.org/10.3390/molecules15031501