3. Experimental
3.1. General
All organometallic reactions were conducted under a nitrogen atmosphere in tetrahydrofuran distilled from sodium benzophenone ketyl immediately before use. Final products were purified on a chromatotron using silica gel-coated rotors (2.0 mm). Hydrobromide and hydrochloride salts of the piperazine products were obtained by using a general procedure [
9,
10] and the salts were crystallized from 95% ethanol. In several cases it was necessary to dilute the ethanolic solution with ether to induce crystallization. Melting points are not corrected.
1H-NMR and
13C-NMR spectra were recorded at 400 MHz and 100 MHz, respectively, on a Bruker Avance insturment.
3.2. Organometallic reagents
n-Butyllithium (2.5 M in hexanes), and
tert-butyllitium (1.7 M in pentane) were commercial reagents. Vinyllithium was generated by the reaction of tetravinyltin with
tert-butyllithium as previously described [
6,
13]. Briefly, a solution of tetravinyltin (0.7 mL, 3.2 mmol) in tetrahydrofuran (10 mL) was treated dropwise at -70 °C for 5 min with
tert-butyllithium (7.4 mL, 12.6 mmol), and the mixture was stirred for an additional 10 min at -70 °C before use. α-Lithiostyrene was generated by dropwise addition of
n-butyllithium (1.2 mL, 3.0 mmol) to a stirred solution of α-bromostyrene (0.43 mL, 3.0 mmol) in tetrahydrofuran (3.0 mL) at -70 °C and the mixture was kept at -70 °C for 5 min before use. Methylmagnesium bromide (3.0 M in diethyl ether), and phenylmagnesium bromide (1.0 M in tetrahydrofuran) were commercial reagents.
2-Chloro-4-vinylpyrimidine (
2). This compound was synthesized as reported by us previously [
7] and characterized as follows (this characterization has not been published in detail previously): an oil, yield 50%;
1H-NMR (CDCl
3): δ 8.57 (d,
J = 5.0 Hz, 1H), 7.22 (d,
J = 5.0 Hz, 1H), 6.70 (m, 1H), 6.52 (m, 1H), 5.80 (m, 1H);
13C-NMR (CDCl
3): δ 165.5, 161.6, 159.9, 133.9, 125.3, 116.5. High-resolution MS (ESI, positive ion mode): calcd. for C
6H
535ClN
2 (M
+ + 1),
m/z 141.0218; found
m/z 141.0220.
2-Chloro-4-(1-phenylvinyl)pyrimidine (3). A solution of α-lithiostyrene (3.0 mmol) in tetrahydrofuran (3 mL) was generated as described above. This solution was treated at once with one portion of a solution of 2-chloropyrimidine (1, 0.21 g, 1.8 mmol) in tetrahydrofuran (2 mL). After stirring for an additional 5 min at -70 °C the mixture was quenched with water (0.1 mL) in tetrahydrofuran (1 mL), and the stirring was continued while the temperature was allowed to rise to 0 °C. After treatment with a solution of 2,3-dichloro-5,6-dicyanoquinone (DDQ, 0.7 g, 3 mmol) in tetrahydrofuran (4 mL) the mixture was stirred at 23 °C for 5 min and then cooled to 0 °C, treated with a cold solution of sodium hydroxide (4 M, 2 mL, 8 mmol) and extracted at 0 °C with hexanes/tetrahydrofuran (1:1, 20 mL). The extract was dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by chromatography eluting with hexanes/dichloromethane (1:1) to provide 3 as colorless oil in 58% yield (0.23 g); 1H-NMR (CDCl3): δ 8.53 (d, J = 5.2 Hz, 1H), 7.40 (m, 3H), 7.31 (m, 2H), 7.09 (d, J = 5.2 Hz, 1H), 6.48 (m, 1H), 5.77 (m, 1H); 13C-NMR (CDCl3): δ 167.7, 161.5, 159.8, 145.9, 138.1, 128.6, 128.5, 128.4, 123.1, 117.5. High-resolution MS (ESI, positive ion mode): calcd. for C12H1035ClN2 (M++ 1), m/z 217.0533; found m/z 217.0526.
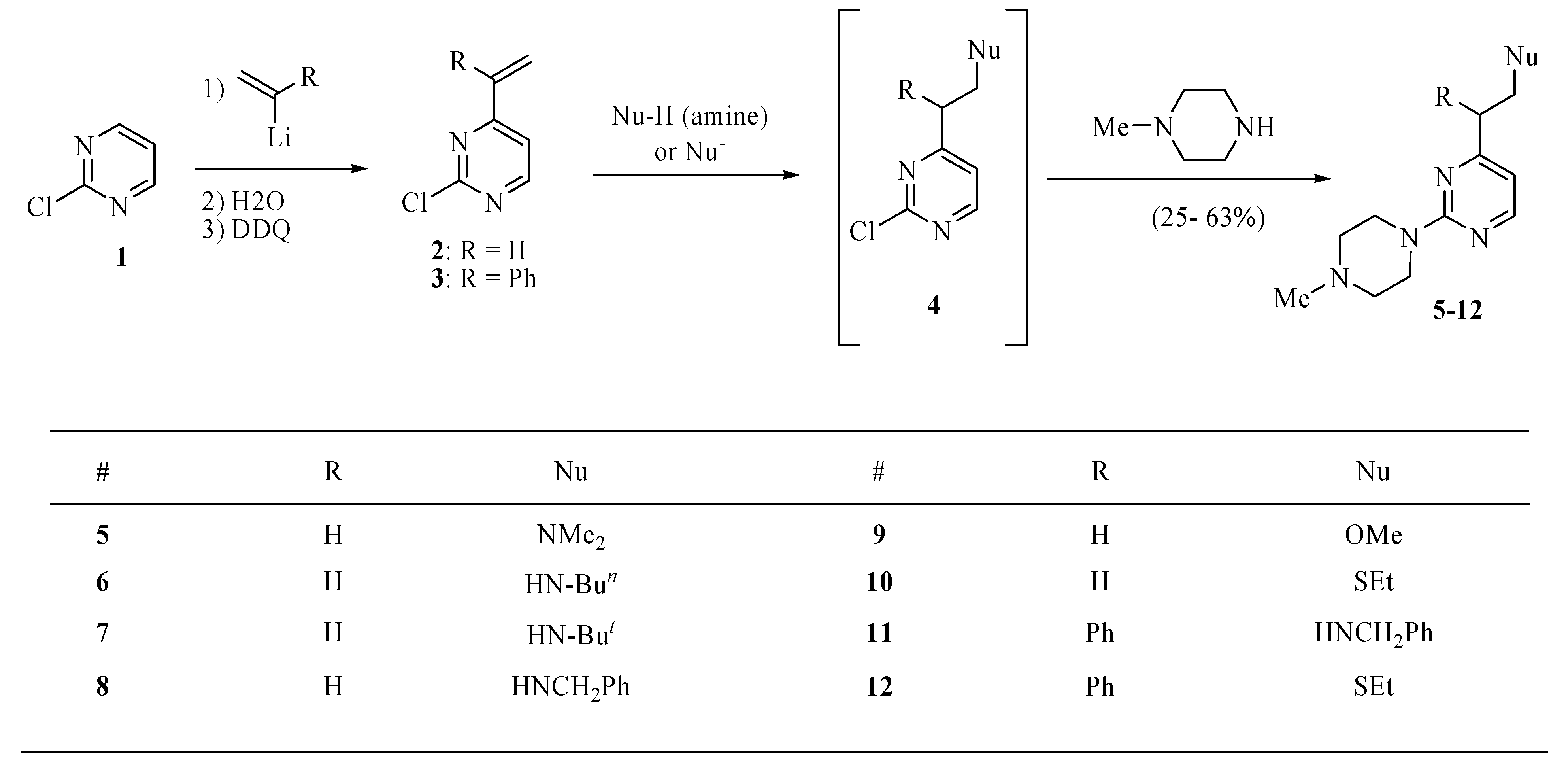
3.3. General procedure for the syntheses of 4 (R = Ph, Nu = OMe, Scheme 1) and 5–12
A solution of
2 or
3 (0.4 mmol) in toluene (3.0 mL) was treated with nucleophile (amine, MeONa, EtSNa, PhSNa, 0.4 mmol). The mixture was stirred at room temperature overnight or at 90 °C for 2 h, then cooled to room temperature, basified with sodium carbonate and extracted with ethyl ether (3 × 15 mL). The extract was dried over magnesium sulfate, and concentrated
in vacuo to provide crude 2-chloro-4-substituted pyrimidines
4 that without purification were used for the reaction with
N-methylpiperazine. Thus, a solution of crude compound
4 and
N-methylpiperazine (0.14 mL, 1.2 mmol) in toluene (4.0 mL) was heated to 80 °C until a TLC analysis on silica gel eluting with hexanes/ethyl ether (4:1) showed the absence of
4 (several hours). The mixture was cooled to room temperature, basified with sodium carbonate and extracted with ethyl ether (3 × 15 mL). The extract was dried over magnesium sulfate, and concentrated under reduced pressure to provide crude compounds
5–12 that were purified by chromatography eluting with hexanes/ethyl ether/methanol (16:3:1). A single compound
4 (R = Ph, Nu = OMe in
Scheme 1) was purified by chromatography in a similar manner.
2-Chloro-4-(2-methoxy-1-phenylethyl)pyrimidine (4, R = Ph, Nu = OMe). This compound was obtained as an oil in 25%; 1H-NMR (CDCl3): δ 8.47 (d, J = 5.2 Hz, 1H), 7.29 (m, 4H), 7.26 (m, 1H), 7.12 (d, J = 5.2 Hz, 1H), 4.31 (dd, J = 8.8, 5.6 Hz, 1H), 4.20 (t, J = 8.8 Hz, 1H), 3.88 (dd, J = 8.8, 5.6 Hz, 1H), 3.35 (s, 3H); 13C-NMR (CDCl3): δ 173.6, 161.3, 159.3, 138.4, 128.9, 128.3, 127.6, 119.5, 74.5, 59.1, 53.2. High-resolution MS (ESI, positive ion mode): calcd. for C13H1335ClN2O (M+ + 1), m/z 249.0795; found m/z 249.0784.
N,N-Dimethyl-2(2-(4-methylpiperazino)pyrimidin-4-yl)ethanamine (5). This compound was obtained as a brown oil in 55% yield; 1H-NMR (CDCl3): δ 8.20 (d, J = 4.8 Hz, 1H), 6.41 (d, J = 4.8 Hz, 1H), 3.86 (t, J = 5.2 Hz, 4H), 2.77–2.74 (m, 2H), 2.71–2.69 (m, 2H), 2.48 (t, J = 5.2 Hz, 4H), 2.35 (s, 3H), 2.31 (s, 6H); 13C-NMR (CDCl3): δ 169.2, 161.7, 157.4, 109.4, 57.9, 54.9, 46.2, 45.2, 43.6, 35.7. High-resolution MS (ESI, positive ion mode): calcd. for C13H23N5 (M+ + 1), m/z 250.2007; found m/z 250.2032.
N-(2-(2-(4-Methylpiperazino)pyrimidin-4-yl)ethyl)butan-1-amine (6). This compound was obtained as an oil in 46% yield; 1H-NMR (CDCl3): δ 8.19 (d, J = 5.0 Hz, 1H), 6.38 (d, J = 5.0 Hz, 1H), 3.84 (t, J = 5.0 Hz, 4H), 2.97 (t, J = 6.6 Hz, 2H), 2.77 (t, J = 6.6 Hz, 2H), 2.63 (t, J = 7.2 Hz, 2H), 2.46 (t, J = 4.8 Hz, 4H), 2.34 (s, 3H), 1.49–1.43 (m, 2H), 0.93–0.89 (m, 2H), 0.91 (t, J = 7.2 Hz, 3H); 13C- NMR (CDCl3): δ 169.4, 161.7, 157.3, 157.1, 109.3, 54.9, 49.5, 48.1, 46.2, 43.6, 37.7, 20.5, 14.0. High-resolution MS (ESI, positive ion mode): calcd. for C15H26N5 (M+ + 1), m/z 278.2340; found m/z 278.2345.
2-Methyl-N-(2-(2-(4-methylpiperazino)pyrimidin-4-yl)ethyl)propan-2-amine (7). This compound was obtained as an oil in 38% yield; 1H NMR (CDCl3): δ 8.20 (d, J = 4.8 Hz, 1H), 6.40 (d, J = 5.0 Hz, 1H), 3.85 (t, J = 3.2 Hz, 4H), 2.97–2.92 (m, 2H), 2.79–2.75 (m, 2H), 2.48 (t, J = 5.0 Hz, 4H), 2.34 (s, 3H), 1.11 (s, 9H); 13C NMR (CDCl3): δ 169.5, 161.6, 157.4, 109.4, 55.0, 50.2, 46.2, 43.6, 40.8, 38.4, 29.0. High-resolution MS (ESI, positive ion mode): calcd. for C15H26 N5 (M+ + 1), m/z 278.2345; found m/z 278.23.
N-Benzyl-2-(2-(4-methylpiperazino)pyrimidin-4-yl)ethanamine tetrahydrobromide (8). This salt was obtained in 62% yield; m.p. 110–112 °C; 1H-NMR (DMSO-d6): δ 10.0 (bs, 1H), 8.96 (bs, 1H), 8.37 (d, J = 4.8 Hz, 1H), 7.58–7.55 (m, 2H), 7.49–7.42 (m, 3H), 6.73 (d, J = 5.4 Hz, 1H), 4.68 (d, J = 14.4 Hz, 2H), 4.24 (t, J = 5.4 Hz, 2H), 3.55–3.50 (m, 2H), 3.39–3.29 (m, 4H), 3.06 (t, J = 7.6 Hz, 4H), 2.86 (bs, 3H); 13C-NMR (CDCl3): δ 171.3, 163.2, 158.3, 143.2, 128.3, 128.1, 125.9, 110.2, 55.3, 54.2, 46.3, 44.0, 39.3, 34.2. MS (ESI) m/z 312 (M+ + H). Anal. Calcd for C18H25N5·4HBr·H2O: C, 33.10; H, 4.78; N, 10.72. Found: C, 33.43; H, 5.10; N, 10.66.
4-(2-Methoxyethyl)-2-4-methylpiperazino)pyrimidine trihydrobromide (9). The free base was obtained as an oil in 55% yield; 1H-NMR for the free base (CDCl3): δ 8.22 (d, J = 5.0 Hz, 1H), 6.44 (d, J = 5.0 Hz, 1H), 3.87 (t, J = 5.0 Hz, 4H), 3.77 (t, J = 6.8 Hz, 2H), 3.37 (s, 3H), 2.85 (t, J = 6.8 Hz, 2H), 2.48 (t, J = 4.8 Hz, 4H), 2.36 (s, 3H); 13C-NMR (CDCl3): δ 168.1, 161.7, 157.4, 109.4, 58.7, 55.0, 46.2, 43.6, 38.0, 29.7. High-resolution MS for the free base (ESI, positive ion mode): calcd. for C12H20 N4O (M+ + 1), m/z 237.1705; found m/z 237.1715. Anal. calcd for: calcd for C12H20N4O·3HBr: C, 30.09; H, 4.24; N, 11.70. Found: C, 29.86; H, 4.34; N, 12.12.
4-(2-(Ethylthio)ethyl)-2-(4-methylpiperazino)pyrimidine (10). This compound was obtained as an oil in 25% yield; 1H-NMR (CDCl3): δ 8.22 (d, J = 5.0 Hz, 1H), 6.41 (d, J = 5.0 Hz, 1H), 3.87 (t, J = 4.8 Hz, 4H), 2.92–2.90 (m, 2H), 2.88–2.86 (m, 2H), 2.60 (q, J = 7.4 Hz, 2H), 2.49 (t, J = 4.8 Hz, 4H), 2.36 (s, 3H), 1.29 (t, J = 7.4 Hz, 3H); 13C-NMR (CDCl3): δ 168.9, 161.7, 157.4, 109.1, 55.0, 46.2, 43.6, 38.0, 29.8, 26.0, 14.7. High-resolution MS (ESI, positive ion mode): calcd. for C13H22N4S (M+ + 1), m/z 267.1632; found m/z 267.1643.
N-Benzyl-2-(2-(4-methylpiperazino)pyrimidin-4-yl)-2-phenylethanamine (11). This compound was obtained as an oil in 57% yield; 1H-NMR (CDCl3) δ 8.13 (d, J = 5.1 Hz, 1H), 7.25 (m, 10H), 6.29 (d, J = 5.1 Hz, 1H), 4.11 (dd, J = 6.4, 8.4Hz, 1H), 3.82 (m, 6H), 3.49 (dd, J = 8.4, 11.8Hz, 1H), 3.07 (dd, J = 6.4, 11.8Hz, 1H), 2.45 (m, 4H), 2.34 (s, 3H); 13C-NMR (CDCl3) δ 170.8, 161.6, 157.7, 141.1, 140.3, 128.6, 128.4, 128.3, 128.1, 127.0, 126.9, 109.7, 55.0, 53.9, 53.1, 53.0, 46.3, 43.7. High resolution MS (ESI, positive ion mode): calcd. for C24H29N5, (M++1), m/z 388.2501, found m/z 388.2491.
4-(2-Ethylthio)-1-phenylethyl)-2-(4-methylpiperazino)pyrimidine (12). This compound was obtained as an oil in 37% yield; 1H-NMR (CDCl3) δ 8.15 (d, J = 5.2 Hz, 1H), 7.29 (m, 5H), 6.34 (d, J = 5.2 Hz, 1H), 4.01 (m, 1H), 3.88 (m, 4H), 3.48 (m, 1H), 3.06 (m, 1H), 2.48 (m, 6H), 2.35 (s, 3H), 1.22 (t, J = 7.2 Hz, 3H); 13C-NMR (CDCl3): δ 170.7, 161.6, 157.7, 141.7, 128.5, 128.1, 127.1, 109.4, 55.0, 53.8, 46.3, 43.7, 36.1, 26.7, 14.8. High-resolution MS (ESI, positive ion mode): calcd. for C19H26N4S (M+ + 1), m/z 343.1956; found m/z 343.1947.
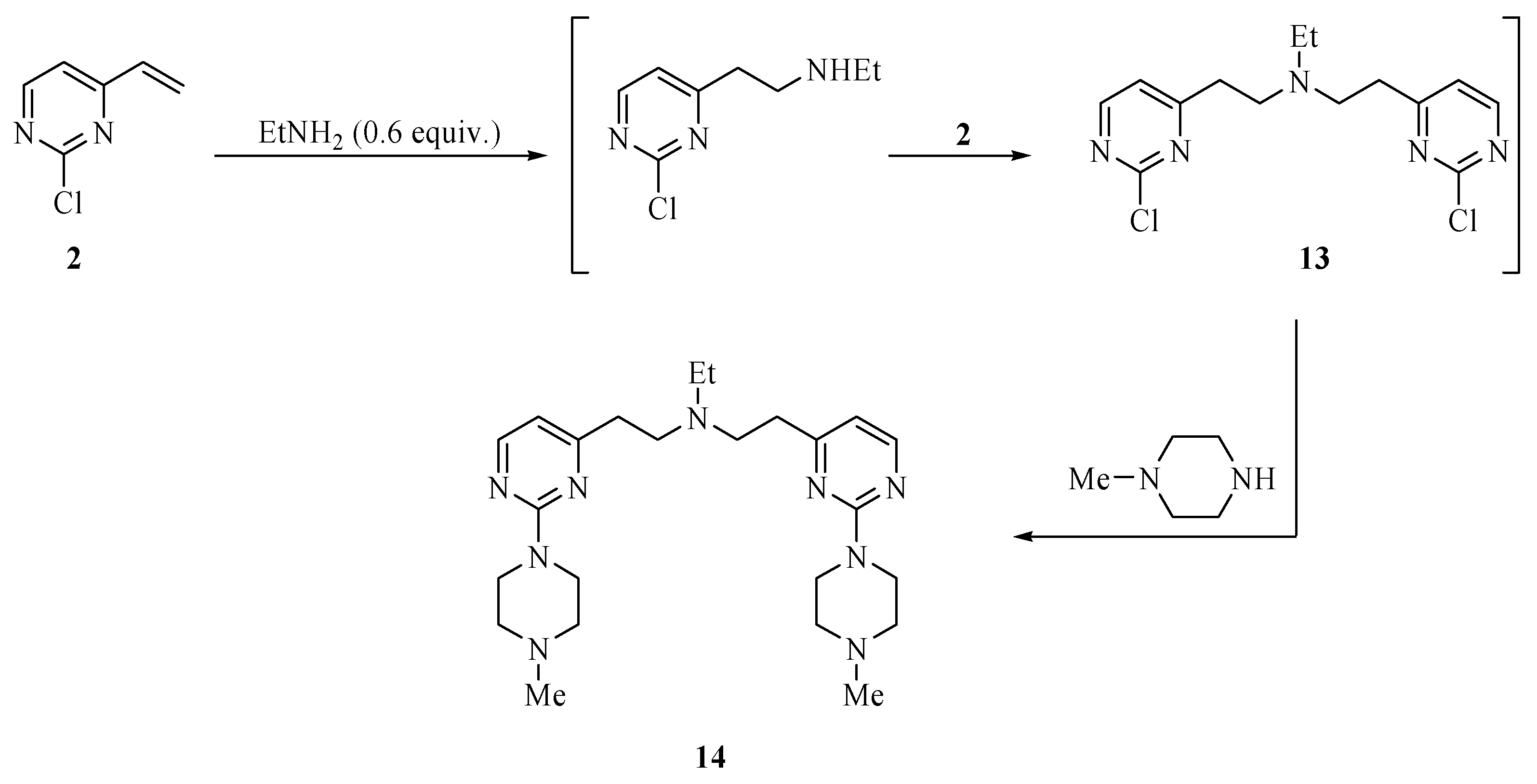
N,N-bis-{[2-(4-Methylpiperazino)pyrimidin-4-yl]ethyl}ethanamine (14). Ethylamine (70% in tetrahydrofuran (0.04 mL, 0.55 mmol) was added to a solution of 2-chloro-4-vinylpyrimidine (2, 0.14 g, 1.0 mmol) in toluene (2.0 mL). The mixture was stirred for 48 h at 23 °C and then heated to 110 °C for 2 h after which time a TLC analysis showed the absence of 2. The mixture was cooled to room temperature, extracted with ethyl ether (2 × 15 mL), dried over magnesium sulfate and concentrated on a rotary evaporator. The residue was purified by chromatography eluting with hexanes/ethyl ether/methanol (8:1:1) to provide 14 as a yellow oil in 38% yield (88 mg); 1H-NMR (CDCl3): δ 8.12 (d, J = 4.8 Hz, 2H), 6.37 (d, J = 4.8 Hz, 2H), 3.87 (t, J = 4.6 Hz, 8H), 2.91 (q, J = 8.0 Hz, 4H), 2.73 (q, J = 8.0 Hz, 4H), 2.64 (q, J = 7.2 Hz, 2H), 2.47 (t, J = 4.6 Hz, 8H), 2.35 (s, 6H), 1.04 (t, J = 7.2 Hz, 3H); 13C -NMR (CDCl3): δ 169.7, 162.3, 157.2, 109.4, 55.0, 51.9, 47.4, 46.3, 43.6, 35.3, 12.1. High-resolution MS (ESI, positive ion mode): calcd. for C24H39N9 (M+ + 1), m/z 454.3405; found m/z 454.3407.
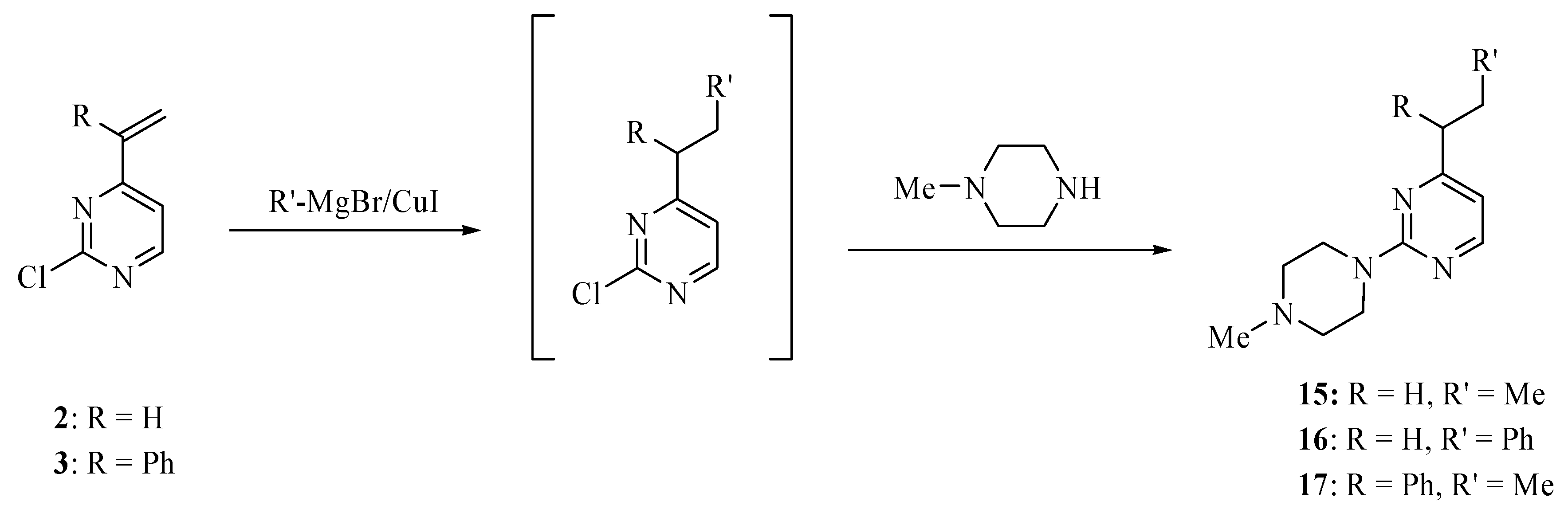
3.4. General procedure for pyrimidines 15–17
Methylmagnesium bromide (3 M in ether, 0.34 mL, 1 mmol) or phenylmagnesium bromide (1 M in tetrahydrofuran, 1.0 mL, 1.0 mmol) was added to cuprous iodide (94 mg, 0.5 mmol) in tetrahydrofuran (2 mL) at -50 °C. The mixture was stirred for 10 min at -50 °C and then treated dropwise with a solution of compound 2 (100 mg, 0.7 mmol or 3 (160 mg, 0.7 mmol) in tetrahydrofuran (3 mL). The mixture was stirred at -40 °C for 2 h, then quenched with aqueous solution of ammonium chloride (2 M, 2.0 mL) and extracted with ethyl acetate (3 × 15 mL). The extract was dried over magnesium sulfate and concentrated on a rotary evaporator. A solution of the residue and N-methylpiperazine (0.12 mL, 1.0 mmol) in toluene (3.0 mL) was heated to 120 °C until a TLC analysis showed the absence of the Grignard adduct (several hours). The mixture was cooled to room temperature, basified with sodium carbonate, and extracted with ethyl ether (3 × 10 mL). The extract was dried over magnesium sulfate and concentrated on a rotary evaporator. The residue was subjected to chromatography eluting with hexanes/ether/methanol (16:3:1) to provide analytically pure products 15–17.
2-(4-Methylpiperazino)-4-propylpyrimidine (15). This compound was obtained as an oil in 56% yield; 1H-NMR (CDCl3): δ 8.22 (d, J = 5.2 Hz, 1H), 6.46 (d, J = 5.2 Hz, 1H), 4.67 (d, J = 14.0 Hz, 2H), 3.96 (t, J = 14.0 Hz, 2H), 3.36–3.25 (m, 7H), 2.56 (t, J = 7.6 Hz, 2H), 1.75–1.70 (m, 2H), 0.97 (t, J = 7.6 Hz, 3H); 13C-NMR (CDCl3): δ 170.2, 160.3, 157.3, 110.0, 65.8, 60.5, 39.8, 38.6, 21.6, 13.8. High-resolution MS (ESI, positive ion mode): calcd. for C12H20 N4 (M+ + 1), m/z 220.1700; found m/z 220.1706.
2-(4-Methylpiperazino)-4-(2-phenylethyl)pyrimidine (16). This compound was obtained as an oil in 77% yield; 1H-NMR (CDCl3): δ 8.19 (d, J = 5.0 Hz, 1H), 7.31 (t, J = 7.2 Hz, 2H), 7.23–7.21 (m, 3H), 6.35 (d, J = 5.0 Hz, 1H), 3.88 (t, J = 4.8 Hz, 4H), 3.04 (q, J = 7.2 Hz, 2H), 2.90 (q, J = 7.2 Hz, 2H), 2.49 (t, J = 4.8 Hz, 4H), 2.37 (s, 3H); 13C-NMR (CDCl3): δ 170.0, 161.7, 157.3, 141.4, 128.4, 128.3, 125.9, 109.0, 55.0, 46.3, 43.6, 39.4, 34.2. High-resolution MS (ESI, positive ion mode): calcd. for C17H22N4 (M+ + 1), m/z 283.1931; found m/z 283.1923.
2-(4-Methylpiperazino)-4-(1-phenylpropyl)pyrimidine (17). This compound was obtained as an oil in 40% yield; 1H-NMR (CDCl3) δ 8.13 (d, J = 5.2 Hz, 1H), 7.30 (m, 4H), 7.20 (m, 1H), 6.32 (d, J = 5.2 Hz, 1H), 3.86 (t, J = 5.2 Hz, 4H), 3.66 (t, J = 7.6 Hz, 1H), 2.46 (t, J = 5.2 Hz, 4H), 2.34 (s, 3H), 2.23 (m, 1H), 1.99 (m, 1H), 0.88 (t, J = 7.6 Hz, 3H); 13C-NMR (CDCl3) δ 172.5, 161.7, 157.4, 142.8, 128.3, 128.2, 126.5, 109.0, 55.4, 55.0, 46.3, 43.7, 27.5, 12.5. High-resolution MS (ESI, positive ion mode): calcd. for C18H24N4 (M + 1)+, m/z 297.2079; found m/z 297.2089.
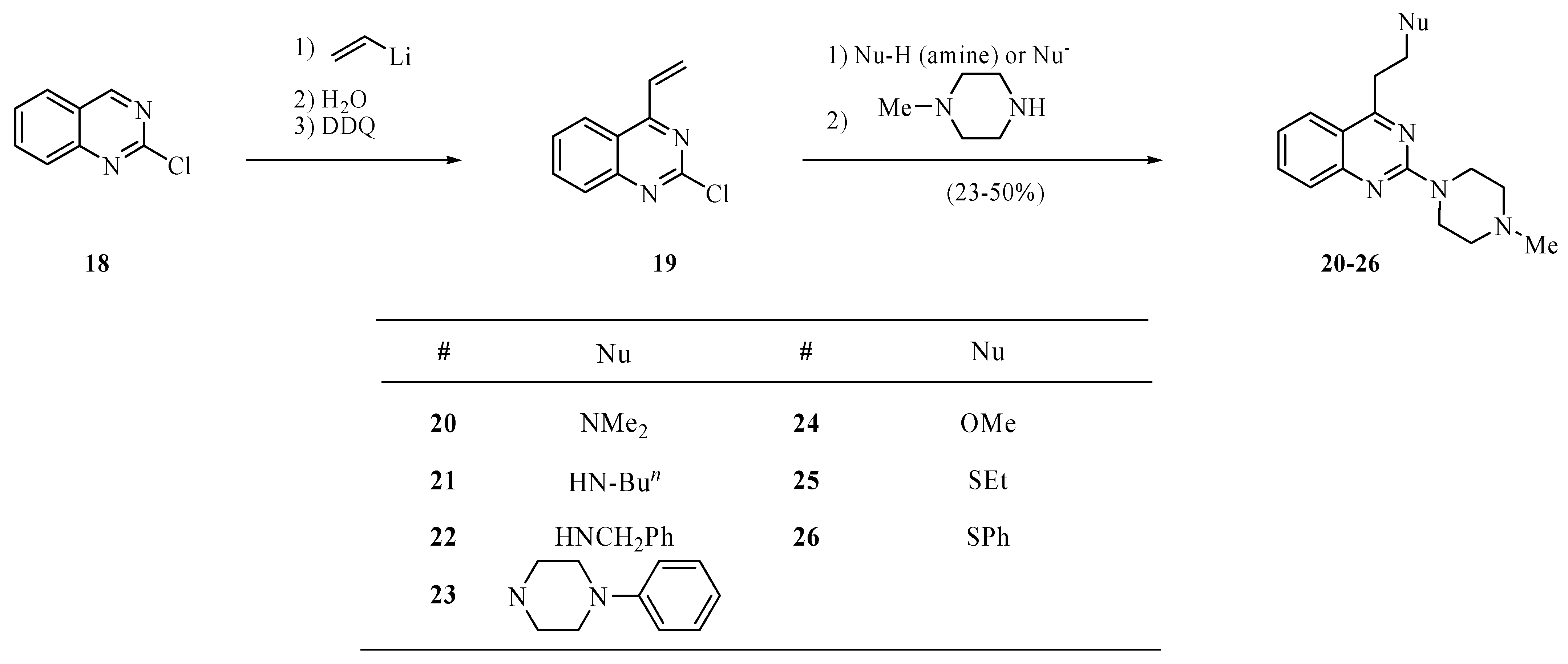
3.5. General procedure for quinazolines 20–26
Synthesis of 2-chloro-4-vinylquinazoline (
19) has been reported by us recently [
7]. To a solution of
19 (190 mg, 1.0 mmol) in toluene (3.0 mL) was added the corresponding nucleophile (amine, MeONa, EtSNa, PhSNa, 1.0 mmol). The mixture was stirred at room temperature overnight or at 90 °C for 2 h, then treated at room temperature with an aqueous solution of ammonium chloride (2M, 2 mL), and extracted with ether (3 × 15 mL). The extract was dried over magnesium sulfate and concentrated under reduced pressure to provide crude 4-substituted 2-chloroquinazoline that without purification was used for the reaction with
N-methylpiperazine. Thus, a solution of the residue after concentration and
N-methylpiperazine (0.30 mL, 2.7 mmol) in toluene (3.0 mL) was heated to 80 °C until a TLC analysis on silica gel eluting with hexanes/ether/methanol (16:3:1) showed the absence of the intermediate product (several hours). The solution was cooled to room temperature, basified with sodium carbonate, and extracted with ethyl ether (3 × 15 mL). The extract was dried over magnesium sulfate and concentrated on a rotary evaporator to provide crude compounds
20–26. These products were purified by chromatography eluting with hexanes/ether/methanol (5:4:1).
N,N-Dimethyl-2-(2-(4-methylpiperazino)quinazolin-4-yl)ethanamine hydrobromide (20). The free base was obtained as a yellow oil in 27% yield; 1H-NMR for the free base (CDCl3): δ 7.86 (d, J = 8.0 Hz, 1H), 7.60–7.57 (m, 2H), 7.18 (t, J = 7.2 Hz, 1H), 3.99 (t, J = 5.0 Hz, 4H), 3.29 (t, J = 7.2 Hz, 2H), 2.85 (t, J = 8.0 Hz, 2H), 2.51 (t, J = 5.0 Hz, 4H), 2.36 (s, 6H), 2.35 (s, 3H); 13C-NMR for the free base (CDCl3): δ 170.1, 158.4, 152.3, 133.3, 126.4, 124.6, 122.1, 118.6, 57.6, 55.1, 46.3, 45.5, 43.8, 32.4. High-resolution MS (ESI, positive ion mode): calcd. for C17H25N5 (M+ + 1), m/z 300.2175; found m/z 300.2188. A hydrobromide salt: m.p. 100–106 °C (dec.). Anal. Calcd. for C17H25N5·3.5HBr·H2O: C, 34.00; H, 5.12; N, 11.66. Found: C, 33.74; H, 5.40; N, 11.26.
N-{2-[2-(4-Methylpiperazino)quinazolin-4-yl]ethyl}butanamine trihydrobromide (21). The free base was obtained as a brown oil in 31% yield; 1H-NMR for the free base (CDCl3): δ 7.83 (d, J = 8.0 Hz, 1H), 7.62–7.53 (m, 2H), 7.14 (d, J = 8.0 Hz, 1H), 3.96 (s, 4H), 3.49 (t, J = 6.6 Hz, 2H), 3.30 (t, J = 6.6 Hz, 2H), 2.81 (t, J = 7.2 Hz, 2H), 2.51 (t, J = 4.8 Hz, 4H), 2.36 (s, 3H), 1.64–1.59 (m, 3H), 1.40–1.32 (m, 2H), 0.90 (t, J = 7.2 Hz, 3H); 13C-NMR for the free base (CDCl3): δ 169.1, 158.3, 152.5, 133.8, 126.6, 124.7, 122.6, 118.6, 55.2, 49.3, 46.8, 46.4, 44.1, 32.5, 31.2, 20.6, 14.0; High-resolution MS (ESI, positive ion mode): calcd. for C19H29N5 (M+ + 1), m/z 328.2503; found m/z 328.2501. A hydrobromide salt: m.p. 153–155 °C. Anal. Calcd. for C19H29N5·3HBr·3H2O: C, 36.04; H, 6.21; N, 11.06. Found: C, 35.89; H, 6.00; N, 11.28.
N-Benzyl-2-(2-(4-methylpiperazino)quinazolin-4-yl)ethanamine trihydrobromide (22). The free base was obtained as a yellow oil in 50% yield; 1H-NMR for the free base (CDCl3): δ 7.84 (d, J = 8.0 Hz, 1H), 7.64–7.56 (m, 2H), 7.33 (d, J = 4.4 Hz, 4H), 7.28–7.25 (m, 1H), 7.18 (t, J = 8.0 Hz, 1H), 3.96 (d, J = 4.6 Hz, 4H), 3.88 (s, 2H), 3.38 (t, J = 6.2 Hz, 2H), 3.22 (t, J = 6.2 Hz, 2H), 2.49 (t, J = 4.6 Hz, 4H), 2.36 (s, 3H), 2.26 (bs, 1H); 13C-NMR for the free base (CDCl3): δ 170.1, 158.4, 152.4, 140.2, 133.6, 128.6, 128.3, 127.2, 126.5, 124.7, 122.4, 118.8, 55.2, 54.2, 46.7, 46.4, 44.0, 33.7. High-resolution MS (ESI, positive ion mode): calcd. for C22H27N5 (M+ + 1), m/z 362.2354; found m/z 362.2345. A hydrobromide salt: m.p. 160–162 °C. Anal. Calcd. for C22H27N5·3HBr: C, 43.73; H, 5.00; N, 11.59. Found: C, 43.88; H, 5.48; N, 11.39.
2-(4-Methylpiperazino)-4-(2-(4-phenylpiperazino)ethyl)quinazoline tetrahydrobromide (23). The free base was obtained as a brown oil in 23% yield; 1H-NMR for the free base (CDCl3): δ 7.90 (d, J = 8.0 Hz, 1H), 7.66–7.58 (m, 2H), 7.31–7.27 (m, 2H), 7.21 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 8.0 Hz, 2H), 6.88 (t, J = 7.2 Hz, 1H), 4.03 (s, 4H), 3.39 (t, J = 7.8 Hz, 2H), 3.27 (t, J = 4.8 Hz, 4H), 3.02 (t, J = 7.8 Hz, 2H), 2.79 (t, J = 4.8 Hz, 4H), 2.55 (t, J = 6.0 Hz, 4H), 2.39 (s, 3H); 13C-NMR for the free base (CDCl3): δ 170.2, 159.3, 152.5, 151.4, 133.6, 129.3, 126.7, 124.8, 122.5, 119.9, 118.8, 116.3, 56.6, 55.2, 53.4, 49.3, 46.3, 43.9, 31.9. High-resolution MS (ESI, positive ion mode): calcd. for C25H32N6 (M+ + 1), m/z 417.2751; found m/z 417.2767. A hydrobromide salt: m.p. 190–194 °C (dec.). Anal. Calcd. for C25H32N6•4HBr•2H2O: C, 38.68; H, 5.19; N, 10.83. Found: C, 38.78; H, 5.51; N, 10.46.
4-(2-Methoxyethyl)-2-(4-methylpiperazino)quinazoline (24). This compound was obtained as a yellow solid in 27% yield; m.p. 35–37 °C; 1H-NMR (CDCl3): δ 7.86 (d, J = 8.0 Hz, 1H), 7.61–7.55 (m, 2H), 7.18 (t, J = 8.0 Hz, 1H), 3.99 (t, J = 4.8 Hz, 4H), 3.94 (t, J = 7.2 Hz, 2H), 3.41–3.38 (m, 5H), 2.51 (t, J = 4.8 Hz, 4H), 2.35 (s, 3H); 13C-NMR (CDCl3): δ 168.9, 158.4, 152.3, 133.4, 126.4, 124.7, 122.2, 118.8, 70.5, 58.8, 55.1, 46.3, 43.8, 34.1. High-resolution MS (ESI, positive ion mode): calcd. for C16H22N4O (M+ + 1), m/z 287.1874; Found m/z 287.1872. Anal. Calcd. for C16H22N4O: C, 67.11; H, 7.74; N, 19.56. Found: C, 67.33; H, 8.05; N, 19.22.
4-(2-(Ethylthio)ethyl)-2-(4-methylpiperazino)quinazoline (25). This compound was obtained as a yellow solid in 35% yield; m.p. 38–40 °C; 1H-NMR (CDCl3): δ 7.83 (d, J = 8.0 Hz, 1H), 7.63–7.55 (m, 2H), 7.18 (t, J = 7.0 Hz, 1H), 4.00 (t, J = 4.8 Hz, 4H), 3.41 (t, J = 7.0 Hz, 2H), 3.07 (t, J = 8.0 Hz, 2H), 2.63 (q, J = 7.0 Hz, 2H), 2.51 (t, J = 4.8 Hz, 4H), 2.36 (s, 3H), 1.30 (t, J = 8.0 Hz, 3H); 13C-NMR (CDCl3): δ 169.8, 158.6, 152.5, 133.6, 126.6, 124.6, 122.4, 118.6, 55.3, 46.5, 44.0, 34.6, 29.3, 26.4, 15.0. High-resolution MS (ESI, positive ion mode): calcd. for C17H24N4S (M+ + 1), m/z 317.1789; Found m/z 317.1800; Anal. Calcd. for C17H24N4S: C, 64.52; H, 7.64; N, 17.70. Found: C, 64.71; H, 7.81; N, 17.30.
2-(4-Methylpiperazino)-4-(2-(phenylthio)ethyl)quinazoline dihydrobromide (26). This compound was obtained as a yellow oil in 50% yield; 1H-NMR for the free base (CDCl3): δ 7.71 (d, J = 8.4 Hz, 1H), 7.62–7.54 (m, 2H), 7.40 (d, J = 7.6 Hz, 2H), 7.30 (t, J = 7.6 Hz, 2H), 7.21 (d, J = 7.2 Hz, 1H), 7.14 (t, J = 6.8 Hz, 1H), 4.00 (s, 4H), 3.51–3.43 (m, 4H), 2.52 (t, J = 5.2 Hz, 4H), 2.37 (s, 3H); 13C-NMR for the free base (CDCl3): δ 169.4, 158.5, 152.5, 136.4, 133.7, 129.6, 129.2, 126.6, 126.4, 124.5, 122.4, 118.6, 55.3, 46.4, 44.0, 33.9, 31.2. High-resolution MS (ESI, positive ion mode): calcd. for C21H24N4S (M+ + 1), m/z 365.1791; Found m/z 365.1800. A hydrobromide salt: m.p. 135–137 °C. Anal. Calcd. for C21H24N4S·2HBr·H2O: C, 46.34; H, 5.18; N, 10.28. Found: C, 46.14; H, 5.18; N, 9.88.

{kind=link}
{kind=link}
{kind=link}
{kind=link}



