Bifunctional Catalysis: Direct Reductive Amination of Aliphatic Ketones with an Iridium-Phosphate Catalyst †

Abstract
:
1. Introduction




2. Results and Discussion
2.1. Optimization of conditions


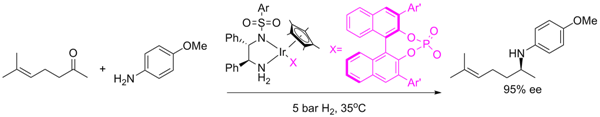{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Ar | X | Addtive | Conv. (%) b | Ee(%) c |
|---|---|---|---|---|---|
| 1 | a | Cl | - | N.R. (17 h) | - |
| 2 | a | Cl | AgSbF6 (2%) | 42 (17 h) | 15 (R) |
| 3 | a | Cl | AgPF6 (2%) | 60 (17 h) | 35 (R) |
| 4 | a | 2-H | - | 11 (17 h) | 47 (R) |
| 5 | a | 2-H | 2 (5%) | 28 (17 h) | 46 (R) |
| 6 | a | 3-H | - | 19 | 84 (S) |
| 7 | a | 3-H | 3 (1%) | 42 | 85 (S) |
| 8 | a | 3-H | 3 (5%) | 60 | 86 (S) |
| 9 | a | 3-H | 3 (8%) | 67 | 89 (S) |
| 10 | a | 3-H | 4Å MS (50 mg) | 30 | 85 (S) |
| 11 | a | 3-H | 4Å MS (100 mg) | 50 | 86 (S) |
| 12 | a | 3-H | 4Å MS | 59 | 86 (S) |
| 13 | a | 3-H | 4Å MS (200 mg) | 59 | 86 (S) |
| 14 | a | 3-H | 3 (1%),4Å MS | 58 | 85 (S) |
| 15 d | a | 3-H | 3 (5%),4Å MS | 63 | 86 (S) |
| 16 | a | 3-H | 3 (8%),4Å MS | 72 | 86 (S) |
| 17 | a | 3-H | 4Å MS | 57 (24 h) | 91 (S) |
| 18 | b | 3-H | - | 5 | - |
| 19 | b | 3-H | 4Å MS | 7 | - |
| 20 | b | 3-H | 3 (8%) | 50 | 71 (S) |
| 21 | b | 3-H | 3 (8%),4Å MS | 60 | 74 (S) |
| 22 | a | 3-H | 4Å MS | >99 (12 h) | 87 (S) |

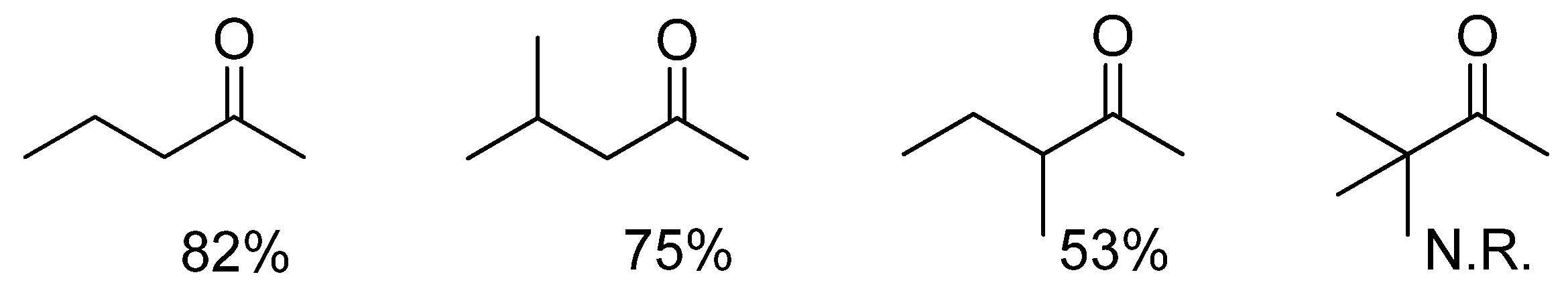
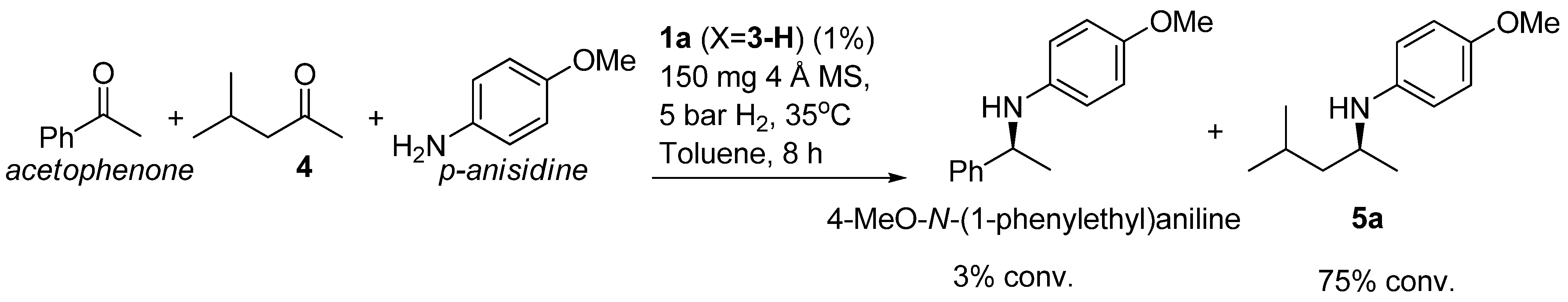
2.2. Scope of substrates
| Entry | Product | Yield (%) | Ee b (%) | Entry | Product | Yield (%) | Ee b (%) |
|---|---|---|---|---|---|---|---|
| 1 |  | 91 | 87 | 7 |  | 80 | 71 |
| 2 |  | 82 | 96 c | 8 |  | 90 | 93 |
| 3 |  | 88 | 90 | 9 |  | 80 | 92 |
| 4 |  | 82 | 93 | 10 |  | 89 | 95 |
| 5 |  | 79 | 91 | 11 |  | 85 | 93 |
| 6 |  | 80 | 49 |
| Entry | Product | Yield (%) | Ee (%) | Entry | Product | Yield (%) | Ee (%) |
|---|---|---|---|---|---|---|---|
| 1 |  | 72 | 80 | 6 |  | 81 | 61 |
| 2 |  | 82 | 96 b | 7 |  | 83 | 64 |
| 3 |  | 86 | 90 | 8 |  | 67 | 82 |
| 4 |  | 77 | 91 | 9 |  | 62 | 82 |
| 5 |  | 85 | 92 | 10 |  | 80 | 91 |
| Entry | Product | Yield (%) | Ee (%) | Entry | Product | Yield (%) | Ee (%) |
|---|---|---|---|---|---|---|---|
| 1 |  | 80 | 88 | 5 |  | 91 | 91 |
| 2 |  | 92 | 94 | 6 |  | 91 | 92 |
| 3 |  | 83 | 95 | 7 |  | 95 | 91 |
| 4 |  | 83 | 92 | 8 b |  | 70 | 84 |

3. Experimental
3.1. General
3.2. General procedure for DARA
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
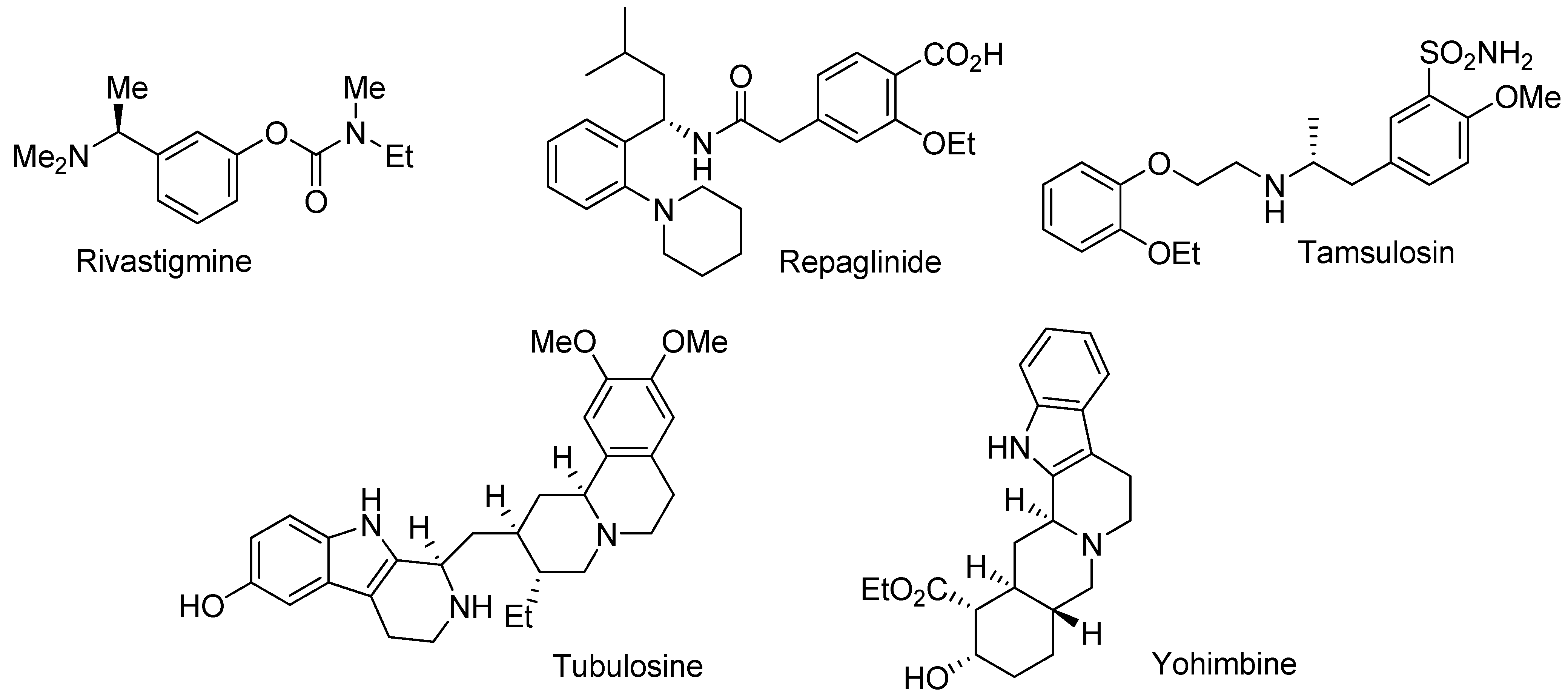
- Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Keβeler, M.; Stürmer, R.; Zelinski, T. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Edit. 2004, 43, 788–824. [Google Scholar] [CrossRef]
- Polinsky, R.J. Clinical pharmacology of rivastigmine: A new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer's disease. Clin. Ther. 1998, 20, 634–647. [Google Scholar] [CrossRef]
- Culy, C.R.; Jarvis, B. Repaglinide - A review of its therapeutic use in type 2 diabetes mellitus. Drugs 2001, 61, 1625–1660. [Google Scholar] [CrossRef]
- Ye, Z.Q.; Lan, R.Z.; Yang, W.M.; Yao, L.F.; Yu, X. Tamsulosin treatment of chronic non-bacterial prostatitis. J. Int. Med. Res. 2008, 36, 244–252. [Google Scholar]
- Ma, W.W.; Anderson, J.E.; McKenzie, A.T.; Byrn, S.R.; McLaughlin, J.L.; Hudson, M.S. Tubulosine - An antitumor constituent of pogonopus-speciosus. J. Nat. Prod. 1990, 53, 1009–1014. [Google Scholar] [CrossRef]
- Bavadekar, S.A.; Ma, G.Y.; Mustafa, S.M.; Moore, B.M.; Miller, D.D.; Feller, D.R. Tethered yohimbine analogs as selective human alpha(2C)-adrenergic receptor ligands. J. Pharmacol. Exp. Ther. 2006, 319, 739–748. [Google Scholar] [CrossRef]
- Blaser, H.U.; Spindler, F. Enantioselective hydrogenation of C=N functions and enamines. In Handbook of Homogeneous Hydrogenation; De Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Volume 3, pp. 1193–1214. [Google Scholar]
- Trifonova, A.; Diesen, J.S.; Chapman, C.J.; Andersson, P.G. Application of phosphine-oxazoline ligands in Ir-catalyzed asymmetric hydrogenation of acyclic aromatic N-arylimines. Org. Lett. 2004, 6, 3825–3827. [Google Scholar]
- Nolin, K.A.; Ahn, R.W.; Toste, F.D. Enantioselective reduction of imines catalyzed by a rhenium(V)-oxo complex. J. Am. Chem. Soc. 2005, 127, 12462–12463. [Google Scholar]
- Moessner, C.; Bolm, C. Diphenylphosphanylsulfoximines as ligands in iridium-catalyzed asymmetric imine hydrogenations. Angew. Chem.-Int. Ed. 2005, 44, 7564–7567. [Google Scholar]
- Yang, Q.; Shang, G.; Gao, W.; Deng, J.; Zhang, X. A highly enantioselective, Pd-TangPhos-catalyzed hydrogenation of N-tosylimines. Angew. Chem.-Int. Ed. 2006, 45, 3832–3835. [Google Scholar]
- Zhu, S.F.; Xie, J.B.; Zhang, Y.Z.; Li, S.; Zhou, Q.L. Well-defined chiral spiro iridium/phosphine-oxazoline cationic complexes for highly enantioselective hydrogenation of imines at ambient pressure. J. Am. Chem. Soc. 2006, 128, 12886–12891. [Google Scholar]
- Reetz, M.T.; Bondarev, O. Mixtures of chiral phosphorous acid diesters and achiral P ligands in the enantio- and diastereoselective hydrogenation of ketimines. Angew. Chem.-Int. Ed. 2007, 46, 4523–4526. [Google Scholar]
- Zhou, H.F.; Li, Z.W.; Wang, Z.J.; Wang, T.L.; Xu, L.J.; He, Y.; Fan, Q.H.; Pan, J.; Gu, L.Q.; Chan, A.S.C. Hydrogenation of quinolines using a recyclable phosphine-free chiral cationic ruthenium catalyst: enhancement of catalyst stability and selectivity in an ionic liquid. Angew. Chem.-Int. Edit. 2008, 47, 8464–8467. [Google Scholar]
- Shirai, S.Y.; Nara, H.; Kayaki, Y.; Ikariya, T. Remarkable positive effect of silver salts on asymmetric hydrogenation of acyclic imines with Cp*Ir complexes bearing chiral N-sulfonylated diamine ligands. Organometallics 2009, 28, 802–809. [Google Scholar]
- Tang, W.J.; Xu, L.J.; Fan, Q.H.; Wang, J.; Fan, B.M.; Zhou, Z.Y.; Lam, K.H.; Chan, A.S.C. Asymmetric hydrogenation of quinoxalines with diphosphinite ligands: A practical synthesis of enantioenriched, substituted tetrahydroquinoxalines. Angew. Chem.-Int. Ed. 2009, 48, 9135–9138. [Google Scholar]
- Tararov, V.I.; Börner, A. Approaching highly enantioselective reductive amination. Synlett 2005, 203–211. [Google Scholar] [CrossRef]
- Tripathi, R.P.; Verma, S.S.; Pandey, J.; Tiwari, V.K. Recent development on catalytic reductive amination and applications. Curr. Org. Chem. 2008, 12, 1093–1115. [Google Scholar] [CrossRef]
- Nugent, T.C.; El-Shazly, M. Chiral amine synthesis - Recent developments and trends for enamide reduction, reductive amination, and imine reduction. Adv. Synth. Catal. 2010. [Google Scholar] [CrossRef]
- Blaser, H.U.; Buser, H.P.; Jalett, H.P.; Pugin, B.; Spindler, F. Iridium ferrocenyl diphosphine catalyzed enantioselective reductive alkylation of a hindered aniline. Synlett 1999, 867–868. [Google Scholar]
- Chi, Y.X.; Zhou, Y.G.; Zhang, X. Highly enantioselective reductive amination of simple aryl ketones catalyzed by Ir-f-Binaphane in the presence of titanium(IV) isopropoxide and iodine. J. Org. Chem. 2003, 68, 4120–4122. [Google Scholar] [CrossRef]
- Kadyrov, R.; Riermeier, T.H. Highly enantioselective hydrogen-transfer reductive amination: Catalytic asymmetric synthesis of primary amines. Angew. Chem.-Int. Ed. 2003, 42, 5472–5474. [Google Scholar]
- Williams, G.D.; Pike, R.A.; Wade, C.E.; Wills, M. A one-pot process for the enantioselective synthesis of amines via reductive amination under transfer hydrogenation conditions. Org. Lett. 2003, 5, 4227–4230. [Google Scholar] [CrossRef]
- Rubio-Pérez, L.; Pérez-Flores, F.J.; Sharma, P.; Velasco, L.; Cabrera, A. Stable preformed chiral palladium catalysts for the one-pot asymmetric reductive amination of ketones. Org. Lett. 2009, 11, 265–268. [Google Scholar]
- Kadyrov, R.; Riermeier, T.H.; Dingerdissen, U.; Tararov, V.; Börner, A. The first highly enantioselective homogeneously catalyzed asymmetric reductive amination: Synthesis of alpha-N-benzylamino acids. J. Org. Chem. 2003, 68, 4067–4070. [Google Scholar]
- Bunlaksananusorn, T.; Rampf, F. A facile one-pot synthesis of chiral beta-amino esters. Synlett 2005, 2682–2684. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.L.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; Zaks, A.; Zhang, T.Y. Key green chemistry research areas - a perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]
- Li, C.Q.; Wang, C.; Villa-Marcos, B.; Xiao, J.L. Chiral counteranion-aided asymmetric hydrogenation of acyclic imines. J. Am. Chem. Soc. 2008, 130, 14450–14451. [Google Scholar]
- Li, C.Q.; Xiao, J.L. Asymmetric hydrogenation of cyclic imines with an ionic Cp*Rh(III) catalyst. J. Am. Chem. Soc. 2008, 130, 13208–13209. [Google Scholar]
- Li, C.Q.; Villa-Marcos, B.; Xiao, J.L. Metal-Brønsted acid cooperative catalysis for asymmetric reductive amination. J. Am. Chem. Soc. 2009, 131, 6967–6969. [Google Scholar]
- Wang, C.; Li, C.Q.; Wu, X.F.; Pettman, A.; Xiao, J.L. pH-regulated asymmetric transfer hydrogenation of quinolines in water. Angew. Chem. -Int. Ed. 2009, 48, 6524–6528. [Google Scholar]
- Klussmann, M. Asymmetric reductive amination by combined Brønsted acid and transition-metal catalysis. Angew. Chem. -Int. Edit. 2009, 48, 7124–7125. [Google Scholar]
- Storer, R.I.; Carrera, D.E.; Ni, Y.; MacMillan, D.W.C. Enantioselective organocatalytic reductive amination. J. Am. Chem. Soc. 2006, 128, 84–86. [Google Scholar]
- Rueping, M.; Sugiono, E.; Azap, C.; Theissmann, T.; Bolte, M. Enantioselective Brønsted acid catalyzed transfer hydrogenation: Organocatalytic reduction of imines. Org. Lett. 2005, 7, 3781–3783. [Google Scholar]
- Hoffmann, S.; Seayad, A.M.; List, B. A powerful Brønsted acid catalyst for the organocatalytic asymmetric transfer hydrogenation of imines. Angew. Chem.-Int. Ed. 2005, 44, 7424–7427. [Google Scholar]
- Bullock, R.M. Catalytic ionic hydrogenations. Chem.-Eur. J. 2004, 10, 2366–2374. [Google Scholar]
- Guan, H.R.; Iimura, M.; Magee, M.P.; Norton, J.R.; Zhu, G. Ruthenium-catalyzed ionic hydrogenation of iminium cations. Scope and mechanism. J. Am. Chem. Soc. 2005, 127, 7805–7814. [Google Scholar]
- Ruano, J.L.G.; Cifuentes, M.M.; Lorente, A.; Ramos, J.H.R. Highly stereoselective reduction of acyclic alpha-sulfinyl ketimines: synthesis of enantiomerically pure beta-aminosulfoxides. Tetrahedron Asymmetry 1999, 10, 4607–4618. [Google Scholar] [CrossRef]
- Hansen, M.C.; Buchwald, S.L. A method for the asymmetric hydrosilylation of N-aryl imines. Org. Lett. 2000, 2, 713–715. [Google Scholar] [CrossRef]
- Liu, X.Y.; Che, C.M. Highly enantioselective synthesis of chiral secondary amines by gold(I)/chiral Brønsted acid catalyzed tandem intermolecular hydroamination and transfer hydrogenation reactions. Org. Lett. 2009, 11, 4204–4207. [Google Scholar] [CrossRef]
- Schnider, P.; Koch, G.; Prétôt, R.; Wang, G.Z.; Bohnen, F.M.; Krüger, C.; Pfaltz, A. Enantioselective hydrogenation of imines with chiral (phosphanodihydrooxazole)iridium catalysts. Chem.-Eur. J. 1997, 3, 887–892. [Google Scholar]
- Cho, B.T.; Chun, Y.S. Enantioselective synthesis of optically-active secondary-amines via asymmetric reduction. Tetrahedron Asymmetry 1992, 3, 1583–1590. [Google Scholar] [CrossRef]
- Mashima, K.; Abe, T.; Tani, K. The half-sandwich hydride and 16-electron complexes of rhodium and iridium containing (1S,2S)-N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine: Relevant to the asymmetric transfer hydrogenation. Chem. Lett. 1998, 1201–1202. [Google Scholar] [CrossRef]
- Mashima, K.; Abe, T.; Tani, K. Asymmetric transfer hydrogenation of ketonic substrates catalyzed by (eta(5)-C5Me5)MCl complexes (M = Rh and Ir) of (1S,2S)-N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine. Chem. Lett. 1998, 1199–1200. [Google Scholar] [CrossRef]
- Miyata, O.; Ishikawa, T.; Ueda, M.; Naito, T. Novel domino elimination-rearrangement-addition reaction of N-alkoxy(arylmethyl)amines to N-alkyl arylamines. Synlett 2006, 2219–2222. [Google Scholar]
- Sato, S.; Sakamoto, T.; Miyazawa, E.; Kikugawa, Y. One-pot reductive amination of aldehydes and ketones with alpha-picoline-borane in methanol, in water, and in neat conditions. Tetrahedron 2004, 60, 7899–7906. [Google Scholar] [CrossRef]
- Kato, H.; Shibata, I.; Yasaka, Y.; Tsunoi, S.; Yasuda, M.; Baba, A. The reductive amination of aldehydes and ketones by catalytic use of dibutylchlorotin hydride complex. Chem. Commun. 2006, 4189–4191. [Google Scholar]
- Duan, H.F.; Sengupta, S.; Petersen, J.L.; Akhmedov, N.G.; Shi, X.D. Triazole-Au(I) complexes: A new class of catalysts with improved thermal stability and reactivity for intermolecular alkyne hydroamination. J. Am. Chem. Soc. 2009, 131, 12100–12102. [Google Scholar]
- Samec, J.S.M.; Bäckvall, J.E. Ruthenium-catalyzed transfer hydrogenation of imines by propan-2-ol in benzene. Chem.-Eur. J. 2002, 8, 2955–2961. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Villa-Marcos, B.; Li, C.; Mulholland, K.R.; Hogan, P.J.; Xiao, J. Bifunctional Catalysis: Direct Reductive Amination of Aliphatic Ketones with an Iridium-Phosphate Catalyst. Molecules 2010, 15, 2453-2472. https://doi.org/10.3390/molecules15042453
Villa-Marcos B, Li C, Mulholland KR, Hogan PJ, Xiao J. Bifunctional Catalysis: Direct Reductive Amination of Aliphatic Ketones with an Iridium-Phosphate Catalyst. Molecules. 2010; 15(4):2453-2472. https://doi.org/10.3390/molecules15042453
Chicago/Turabian StyleVilla-Marcos, Barbara, Chaoqun Li, Keith R. Mulholland, Philip J. Hogan, and Jianliang Xiao. 2010. "Bifunctional Catalysis: Direct Reductive Amination of Aliphatic Ketones with an Iridium-Phosphate Catalyst" Molecules 15, no. 4: 2453-2472. https://doi.org/10.3390/molecules15042453
APA StyleVilla-Marcos, B., Li, C., Mulholland, K. R., Hogan, P. J., & Xiao, J. (2010). Bifunctional Catalysis: Direct Reductive Amination of Aliphatic Ketones with an Iridium-Phosphate Catalyst. Molecules, 15(4), 2453-2472. https://doi.org/10.3390/molecules15042453