2. 1,3-Dienes
The Pd(0) catalyzed coupling of 1-alkoxy-1,3-dienes, obtained by the reaction of α,β-unsaturated acetals with superbase LIC-KOR (equimolar mixture of
n-butyllithium and potassium
tert-butoxide) in the presence of the suitable electrophile (
Scheme 2), has been studied in our laboratory [
12].
Scheme 2.
Reaction of α,β-unsaturated acetals with superbase LIC-KOR.
Scheme 2.
Reaction of α,β-unsaturated acetals with superbase LIC-KOR.
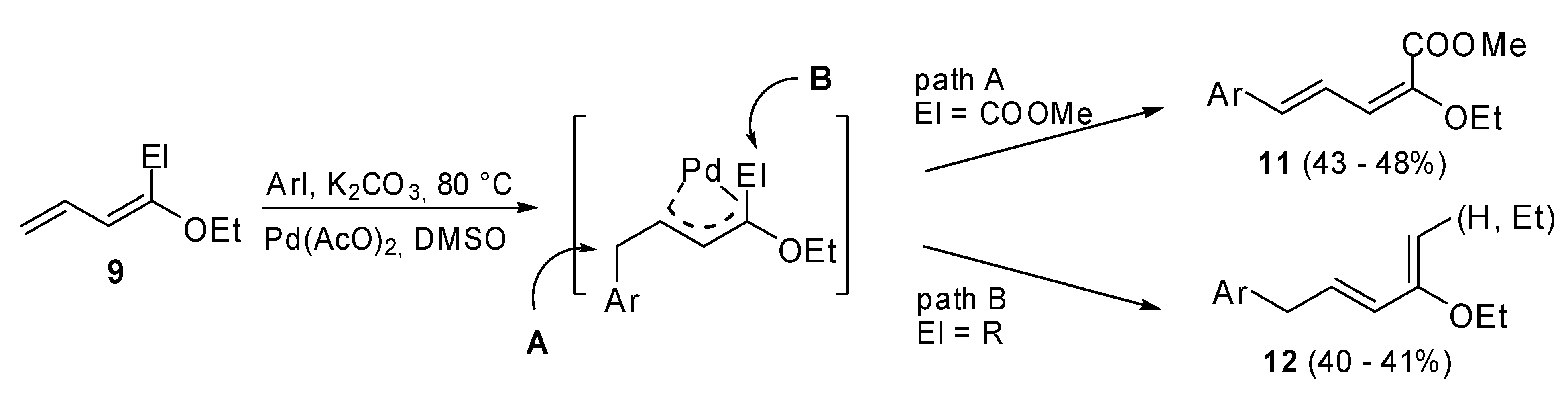
When derivatives
9 were coupled with aryl halides
, two different pathways (
Scheme 3) were observed. The choice of pathway depends on the group bonded to the C
1 of the dienic moiety. In the case of
(E)-methyl 2-ethoxypenta-2,4-dienoate (El = COOMe), the corresponding 4-aryl derivatives
11 were isolated in a regio- and stereoselective manner (path A,
Scheme 3). On the contrary, in the case of 1-alkyl-1-alkoxydienes (El = Me,
n-Pr) arylated dienes
12, that are isomers of the expected dienes, were synthesized (path B,
Scheme 3). A common π-allylpalladium intermediate which undergoes a β-hydride elimination process from two different sites was hypothesized. Pathway B was probably favored for steric reasons.
Scheme 3.
Heck reaction on 1-ethoxy-buta-1,3-dienes.
Scheme 3.
Heck reaction on 1-ethoxy-buta-1,3-dienes.
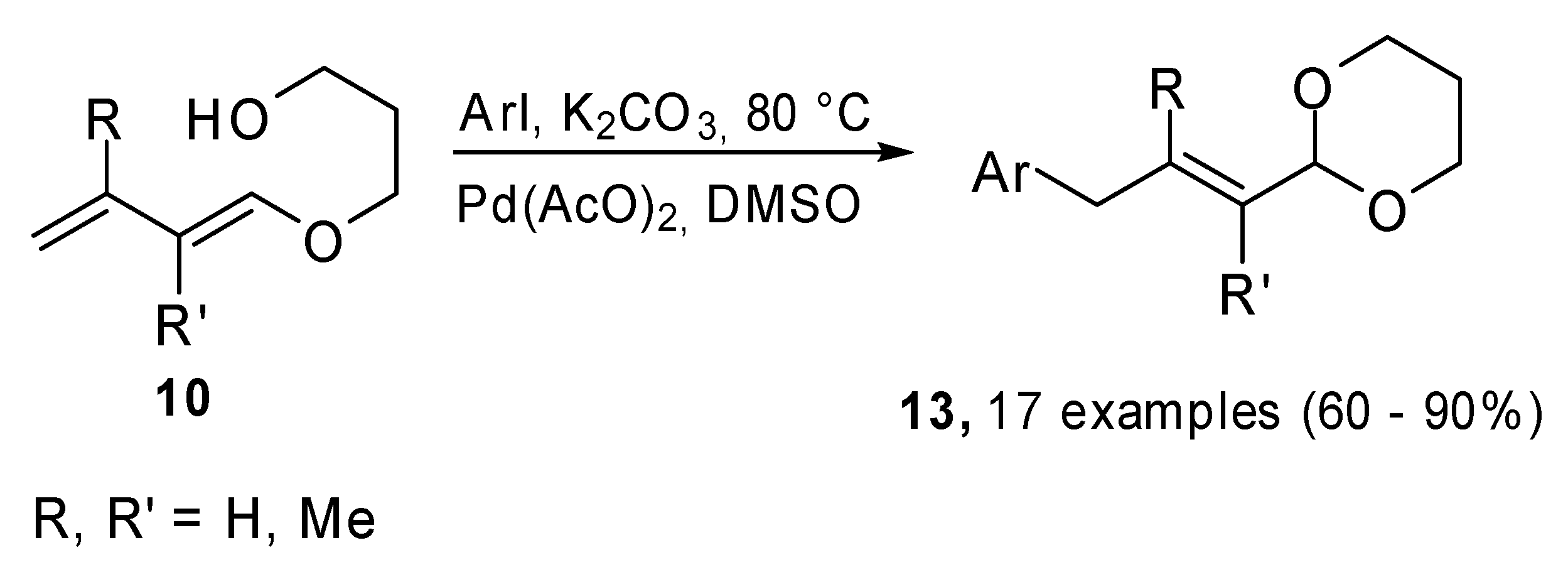
When the Heck cross coupling was carried out on compounds
10, the reaction followed a different pathway because of the presence of the nucleophilic hydroxyl group in the substrate (
Scheme 4). The 1,3-dioxane ring was reformed and the process formally led to a γ-arylation process on the α,β-unsaturated protected carbonyl compounds.
Scheme 4.
Heck reaction on 1-(3-hydropropoxy)buta-1,3-dienes with aryl iodides.
Scheme 4.
Heck reaction on 1-(3-hydropropoxy)buta-1,3-dienes with aryl iodides.
This methodology was extended to arendiazonium salts, because the Heck reaction on these substrates is mild and fast. Moreover, their use is synthetically convenient in comparison to aryl halides since many of them, especially iodides, are prepared from diazonium salts [
13]. The couplings were carried out in anhydrous MeCN at room temperature in the presence of NaAcO as the base and Pd(AcO)
2 as the catalyst. The reactions showed total regio- and stereoselectivity, unfortunately the coupling yields were modest, probably they were affected by the stability of either the diazonium salt or the dienes under the reaction conditions.
Scheme 5.
Heck reaction on 1-(3-hydropropoxy)buta-1,3-dienes with arendiazonium salts.
Scheme 5.
Heck reaction on 1-(3-hydropropoxy)buta-1,3-dienes with arendiazonium salts.
As the yields of the Heck coupling between 1-ethoxybuta-1,3-dienes
9 and aryl halides described in
Scheme 3 were not satisfactory, different reaction conditions were tried in our laboratory. In particular the effect of the addition of ionic liquid to the reactive mixture was evaluated and it was observed that the presence of TBAB increased both the yields and the reaction rates (
Scheme 6) [
14]. The best results were obtained using a mixture of TBAB and DMSO as the solvent, NaAcO as the base and Pd(AcO)
2 as the catalyst. However the subsequent couplings were carried out in pure TBAB, as the possibility of developing a solvent free method was, in our opinion, more significant than the longer reaction times it caused (4 h instead of 2 h). Moreover the Heck reaction was successfully applied to aryl bromides.
Scheme 6.
Heck reaction on 1-ethoxy-buta-1,3-dienes in pure TBAB.
Scheme 6.
Heck reaction on 1-ethoxy-buta-1,3-dienes in pure TBAB.
(
Z)-Buta-1,3-dien-1-yl nonaflate (
14) was obtained through aldehyde-free pathways, exploiting the lithiation of 2,5-dihydrofuran followed by the cyclo fragmentation of the metallated heterocycle by Reissig and coworkers (
Scheme 7).
Scheme 7.
Synthesis of (Z)-buta-1,3-dien-1-yl nonaflate by lithiation of 2,5-dihydrofuran.
Scheme 7.
Synthesis of (Z)-buta-1,3-dien-1-yl nonaflate by lithiation of 2,5-dihydrofuran.
The so obtained conjugated diene
14 was found to react with monosubstituted alkenes under phosphine free conditions and using Pd(OAc)
2 as the catalyst. It was observed that the coupling was influenced by the presence of lithium chloride as the co-catalyst, the product (3
E, 5
Z)-octa-3,5,7-trien-2-one (
15) was obtained in good yields and high (
E)-selectivity. In fact as expected, nonaflate
14 seemed to react while retaining its configuration as illustrated in
Scheme 8 [
15].
Scheme 8.
Heck reaction of (Z)-buta-1,3-dien-1-yl nonaflate.
Scheme 8.
Heck reaction of (Z)-buta-1,3-dien-1-yl nonaflate.
Li and coworkers reported an interesting example of Heck coupling between vinyl chlorides
16,
18 and
21 and different olefins (
Scheme 9) [
16]. In particular, they demonstrated that the vinylic C-Cl bond is activated upon complexation with Fe(CO)
3. The diene was firstly and easily bonded to Fe(CO)
3 and then reacted under the Heck conditions. In consideration of the ready decomplexation of the diene−Fe(CO)
3 under mild conditions, this method could find many applications in organic synthesis.
Scheme 9.
Heck reactions of vinyl chlorides and diene−Fe(CO)3 complexes.
Scheme 9.
Heck reactions of vinyl chlorides and diene−Fe(CO)3 complexes.
3. 1,2-Dienes
As previously outlined, allenes are reactive substrates in Pd(0) catalyzed reactions and, in recent years, much work has been done to find new synthetic methods which utilize these molecules for the preparation of structurally complicated products.
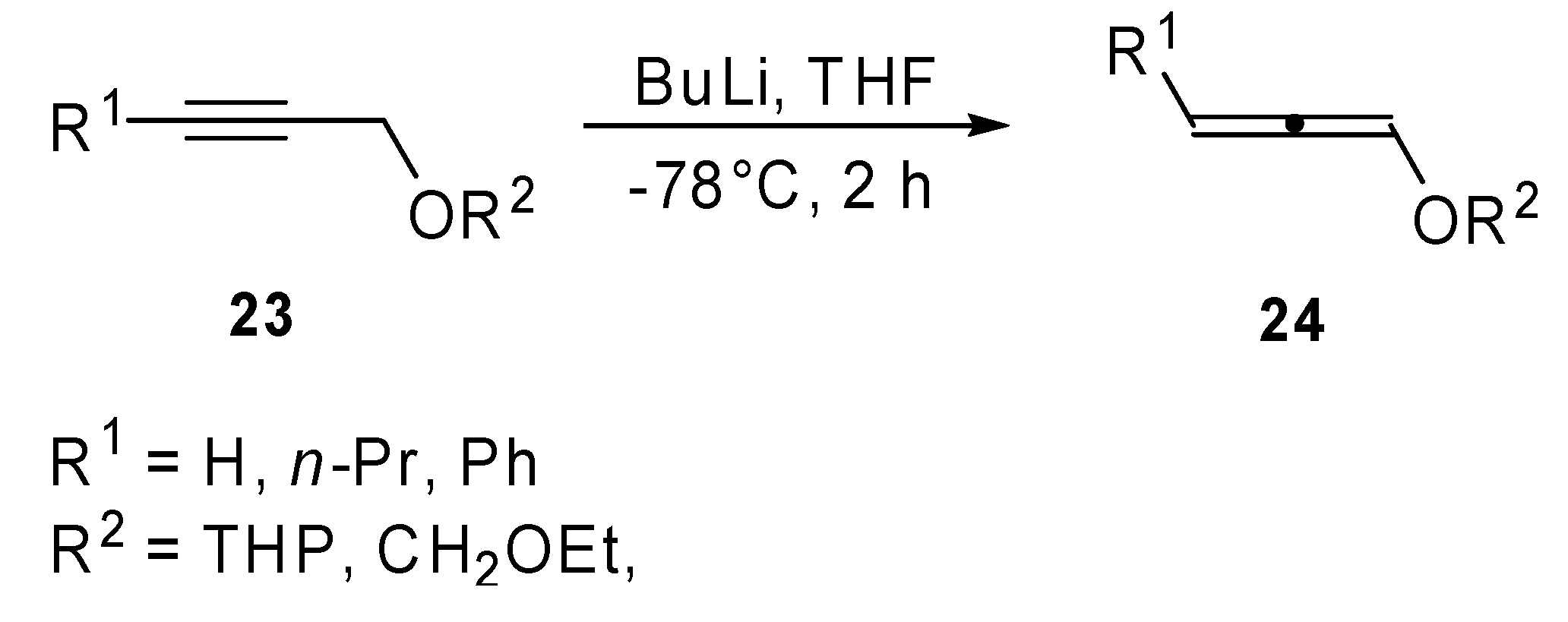
Recently, in our laboratory some attention has been paid to the reactivity of protected 1,2-dien-1-ols in the Heck reaction conditions. A new synthetic method for the preparation of α-arylated α,β-unsaturated aldehydes (
26) has been proposed [
17]. Protected dienols
24 were prepared from the corresponding alkynes
23 by reaction with butyllithium (
Scheme 10). As has been shown in literature, a mixture of propargyl and allenyl ethers was obtained in the case of internal alkynes.
Scheme 10.
Isomerization reaction of alkynes to allenes in the presence of BuLi.
Scheme 10.
Isomerization reaction of alkynes to allenes in the presence of BuLi.
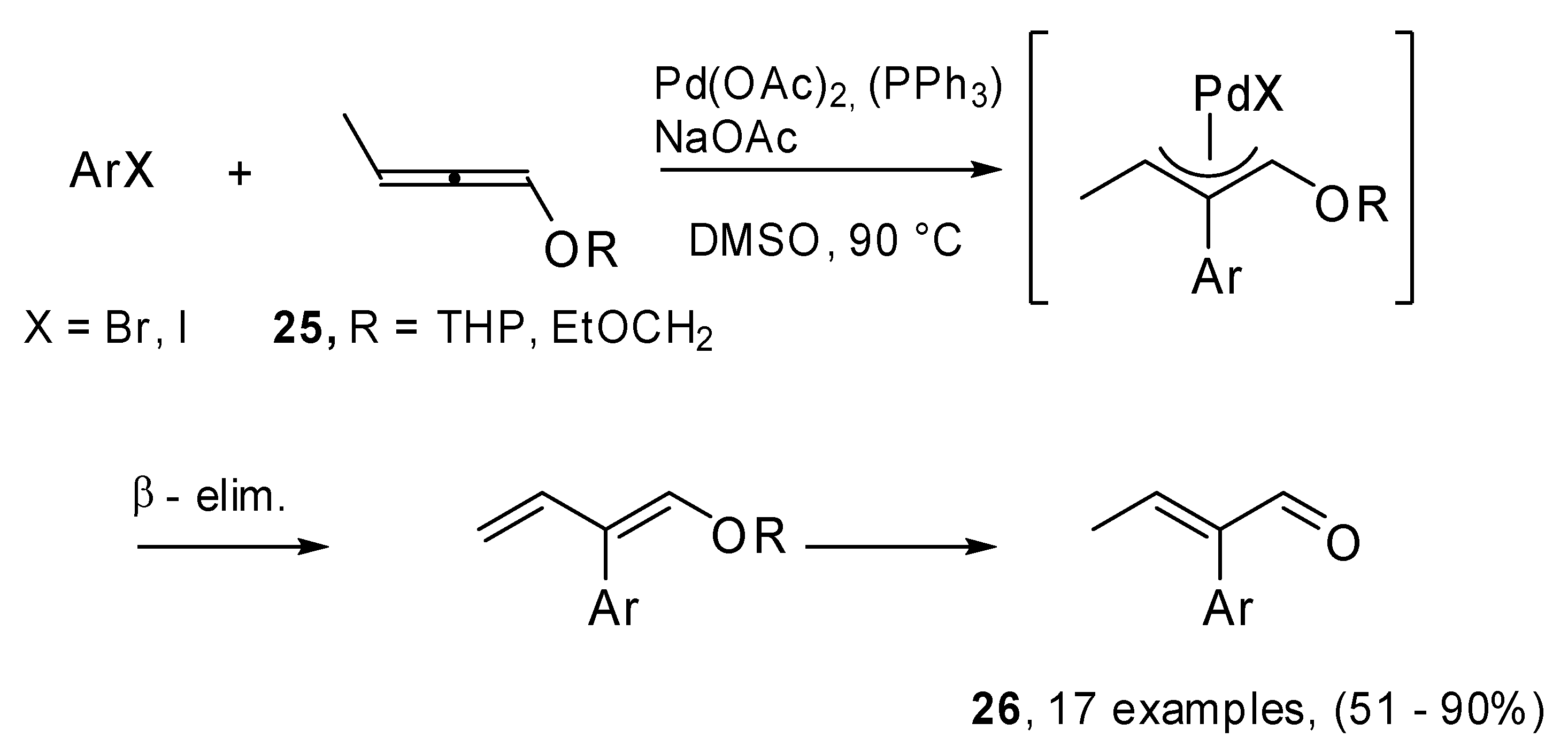
The methodology was applied to differently substituted iodo- and bromoarenes (
Scheme 11), in DMSO at 90 °C. NaAcO was used as the base and PPh
3 was added in the case of bromo derivatives. Seventeen examples were reported. The reactions were regio- and stereoselective, furthermore they turned out to be strongly influenced by substituent steric effects. Moreover, in the case of bromoarenes electronic effects appeared to be important.
Scheme 11.
Heck couplings of aryl halides and allenols to afford α-arylated α,β-unsaturated aldehydes.
Scheme 11.
Heck couplings of aryl halides and allenols to afford α-arylated α,β-unsaturated aldehydes.
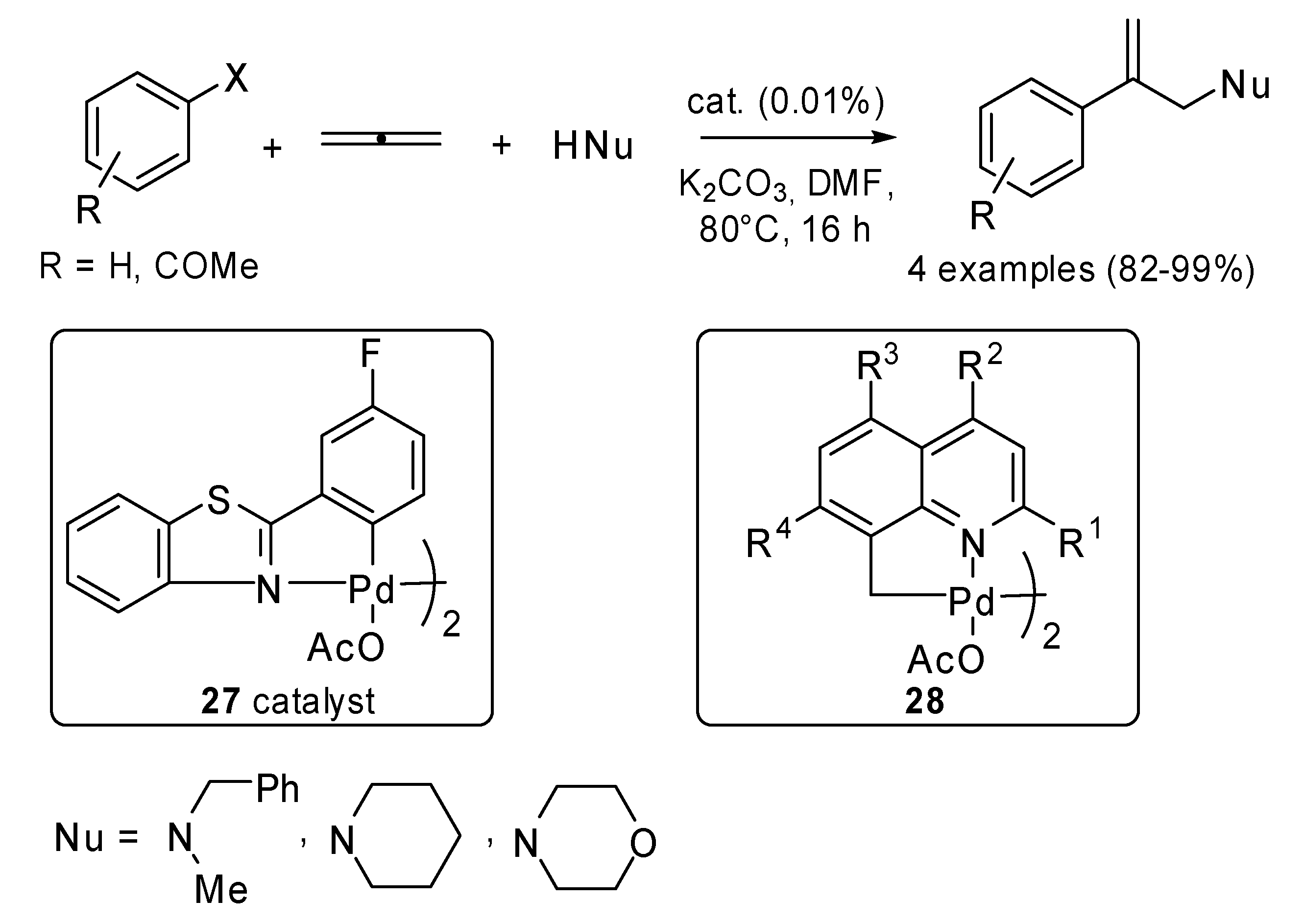
Grigg and coworkers did a lot of work in studying the reactivity of allenes in Pd(0) catalyzed reactions. The use of non-phosphine palladacycles
27 as efficient catalysts in Heck reaction was applied to a three-component cascade process involving haloarenes, 1,2-propene and different amines (
Scheme 12) [
18]. Some non-phosphine 8-methyl quinoline based dimeric palladacycles
28, which possess an sp
3–C bond, were tested too. They proved to be efficient pre-catalysts for both Heck and 3-component cascade processes [
19].
Scheme 12.
Three-component cascade process involving haloarenes, 1,2-propene and different amines.
Scheme 12.
Three-component cascade process involving haloarenes, 1,2-propene and different amines.
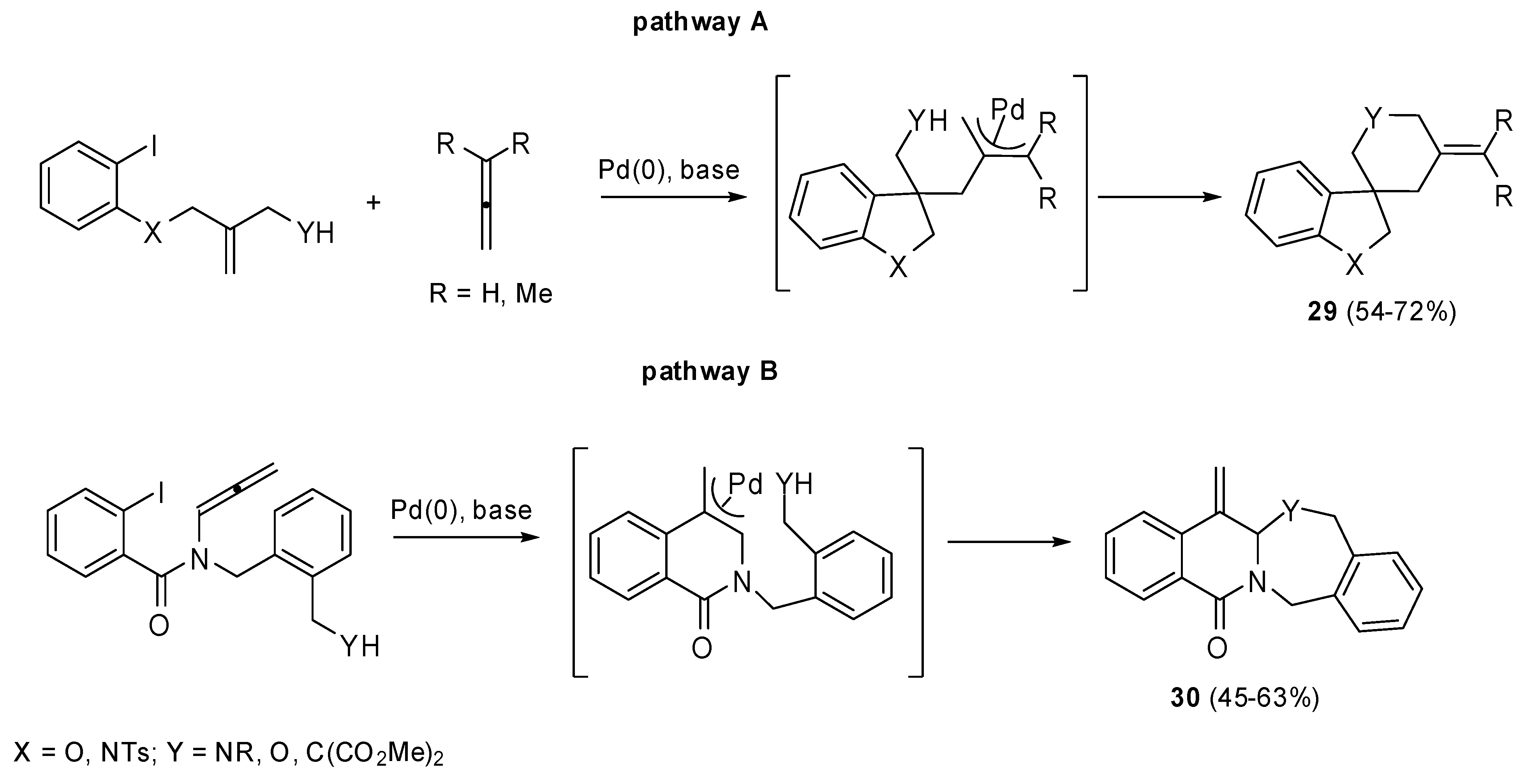
Some palladium catalyzed carbo- and heteroannulation allene cascade reactions, which allowed the synthesis of spiro- and fused heterocycles, were proposed by the same group (
Scheme 13) [
20].
The oxidative process can be followed by an
exo-trig cyclization, the intermolecular allene insertion and the intramolecular capture of the resultant (π-allyl)-palladium complex with a tethered nucleophile (pathway A,
Scheme 13) affords the spiro-fused ring system
29. Otherwise the (π-allyl)-palladium complex is generated by an
exo-dig cyclization of the Ar-Pd species onto the adjacent 1,2-dienamide. The subsequent attack by a nucleophile permits the formation of bicyclic lactams
30 (pathway B,
Scheme 13).
Scheme 13.
Synthesis of spiro- and fused heterocycles by Heck reaction.
Scheme 13.
Synthesis of spiro- and fused heterocycles by Heck reaction.
Moreover an intramolecular Heck reaction between aryl/heteroaryl iodides and alkylallenes was exploited to prepare functionalized 1,3-dienes (
Scheme 14). These were subsequently reacted with various dienophiles to give the corresponding Diels–Alder adducts
31 [
21].
Scheme 14.
Three component Heck–Diels–Alder cascade process.
Scheme 14.
Three component Heck–Diels–Alder cascade process.
Optimum reaction conditions were found to depend on the substituents present in the aryl iodide. It was necessary to work at 120 °C for 48 h to get good yields in the case of electron withdrawing groups, whereas the best results were obtained at 90 °C, when electron donating groups were present on the aromatic ring. This is possibly due to the reduced stability of the ArPdI complex.
A three component palladium-indium mediated diastereoselective cascade allylation of imines with allenes and aryl iodides was proposed by Grigg and coworkers too [
22]. The synthesis provided
N-tosyl and
N-aryl homoallyl amines
32. Furthermore the use of enantiomerically pure
N-
tert-butanesulfinyl imines afforded the desired products with excellent diastereoselectivity. Firstly isatin imines were explored (
Scheme 15), in this case a spiro-oxindole was obtained.
Scheme 15.
Cascade allylation of isatin imines.
Scheme 15.
Cascade allylation of isatin imines.
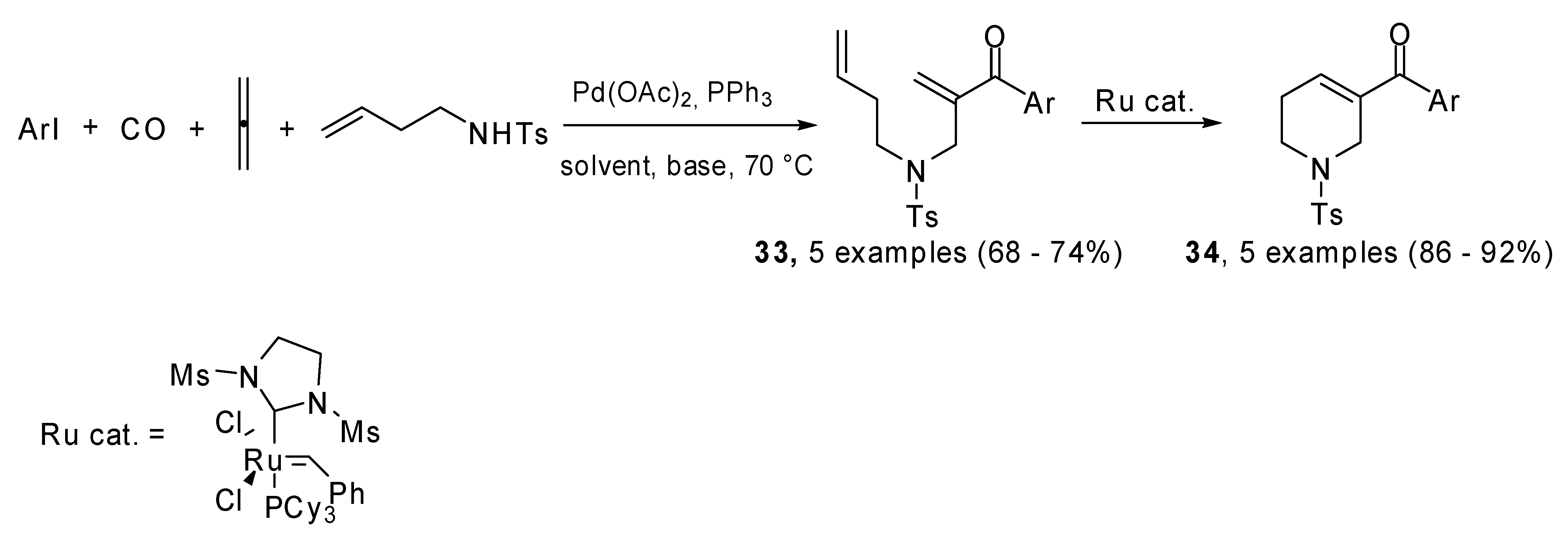
1,6-Dienones and 1,7-dienones (
33) were prepared using a four-component Pd(0) catalyzed four component process which involved carbon monoxide, allene and heteroaryl iodides. They generated a (π-allyl)-palladium species which reacted with alkene tethered nitrogen nucleophiles. A subsequent ring closing metathesis produced five- and six-membered
N-heterocyclic enones
34 which are active dipolarophiles in 1,3-dipolar cycloaddition reactions (
Scheme 16) [
23].
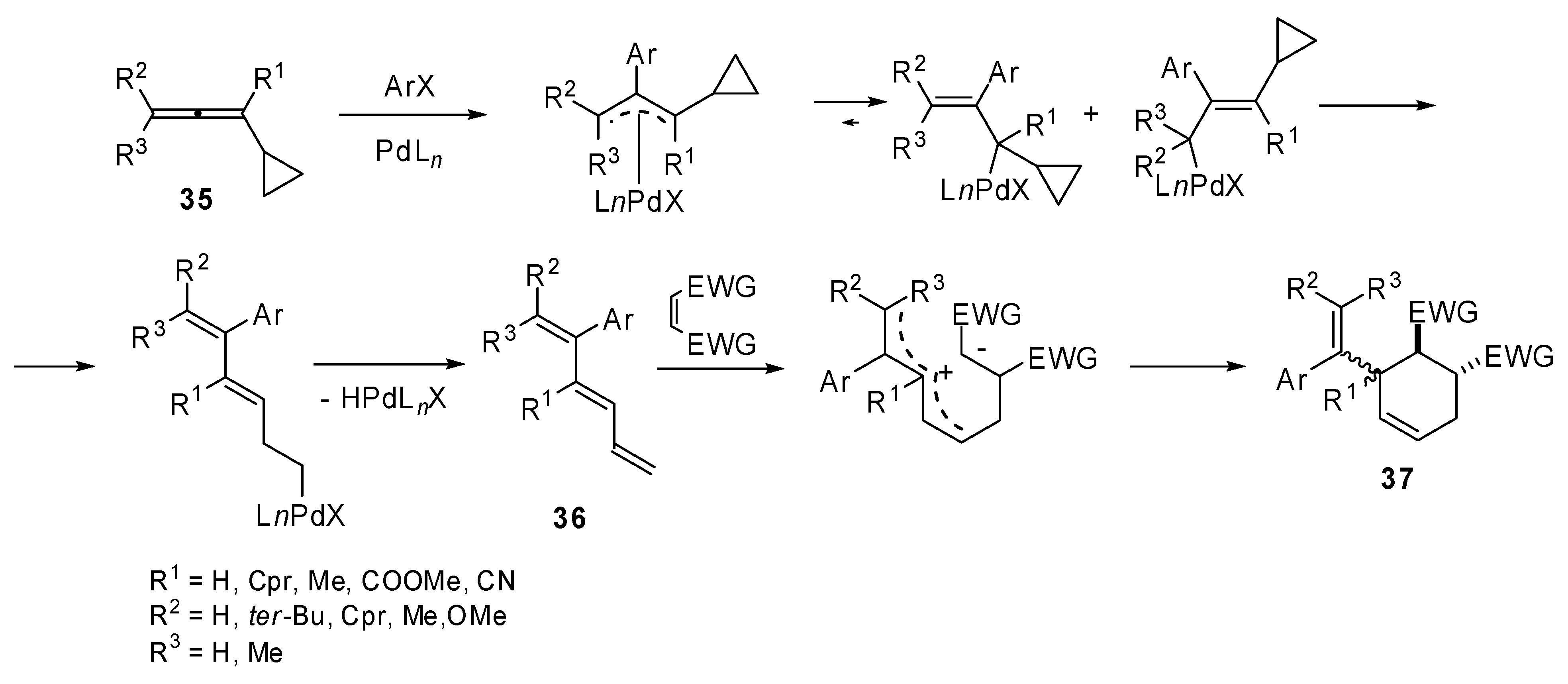
The original studies on the use of cyclopropylallenes in cascade reactions involving palladium catalyzed coupling were carried out by de Meijere
et al. 1,3-Dicyclopropyl-1,2-propadiene (
35) was coupled with several aryl iodides and bromides under palladium catalysis in the presence of a dienophile which underwent a domino Heck–Diels–Alder reaction. 3-(1’-Arylalkenyl)-substituted cyclohexenes
37 were achieved in moderate to good yields [
24]. Then the methodology was extended to differently substituted cyclopropylallenes [
25]. When the reaction was performed without the added dienophile, the intermediate coupling product
36 was isolated and characterized, unfortunately it underwent polymerization within a few hours at room temperature. Moreover the [4 + 2] cycloaddition was found to proceed in a non-concerted fashion (
Scheme 17).
Scheme 16.
Four-component Pd(0) catalyzed cascade/ring closing metathesis.
Scheme 16.
Four-component Pd(0) catalyzed cascade/ring closing metathesis.
Scheme 17.
Domino Heck Diels-Alder reaction by substituted 1,3-dicyclopropyl-1,2-propadienes.
Scheme 17.
Domino Heck Diels-Alder reaction by substituted 1,3-dicyclopropyl-1,2-propadienes.
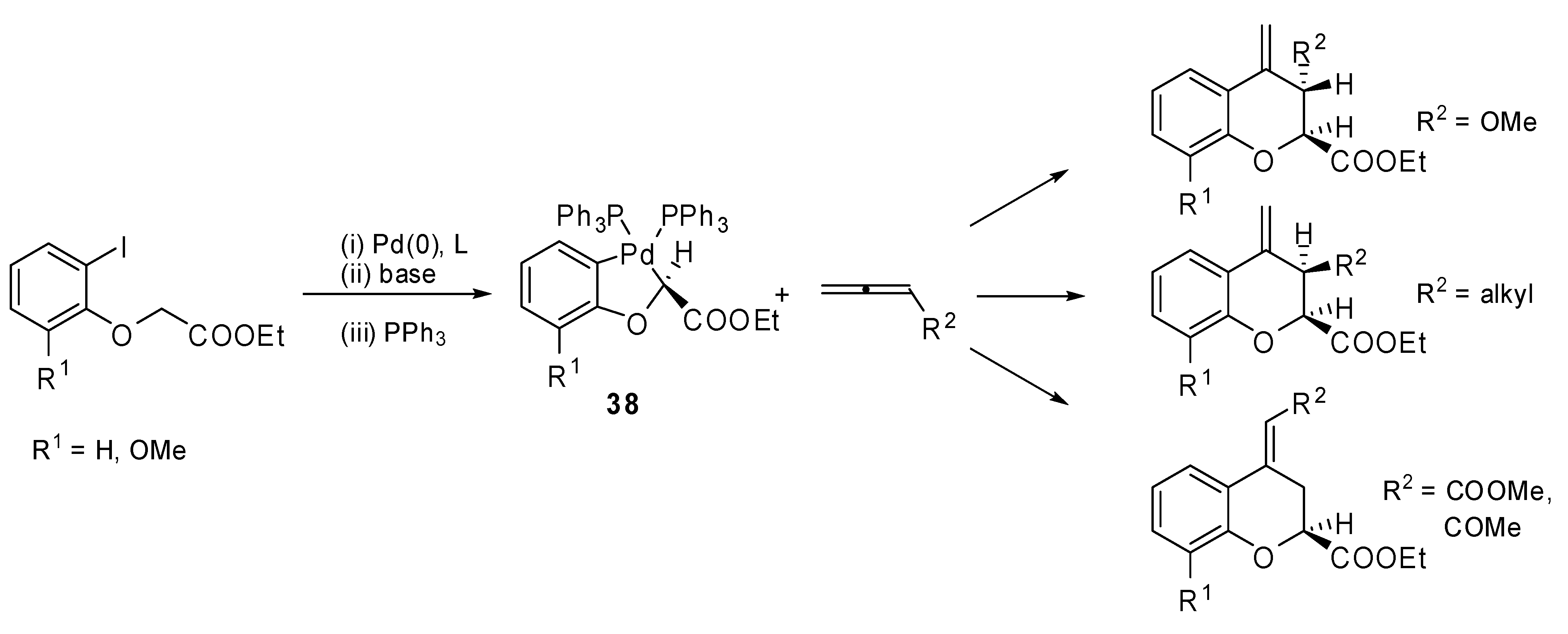
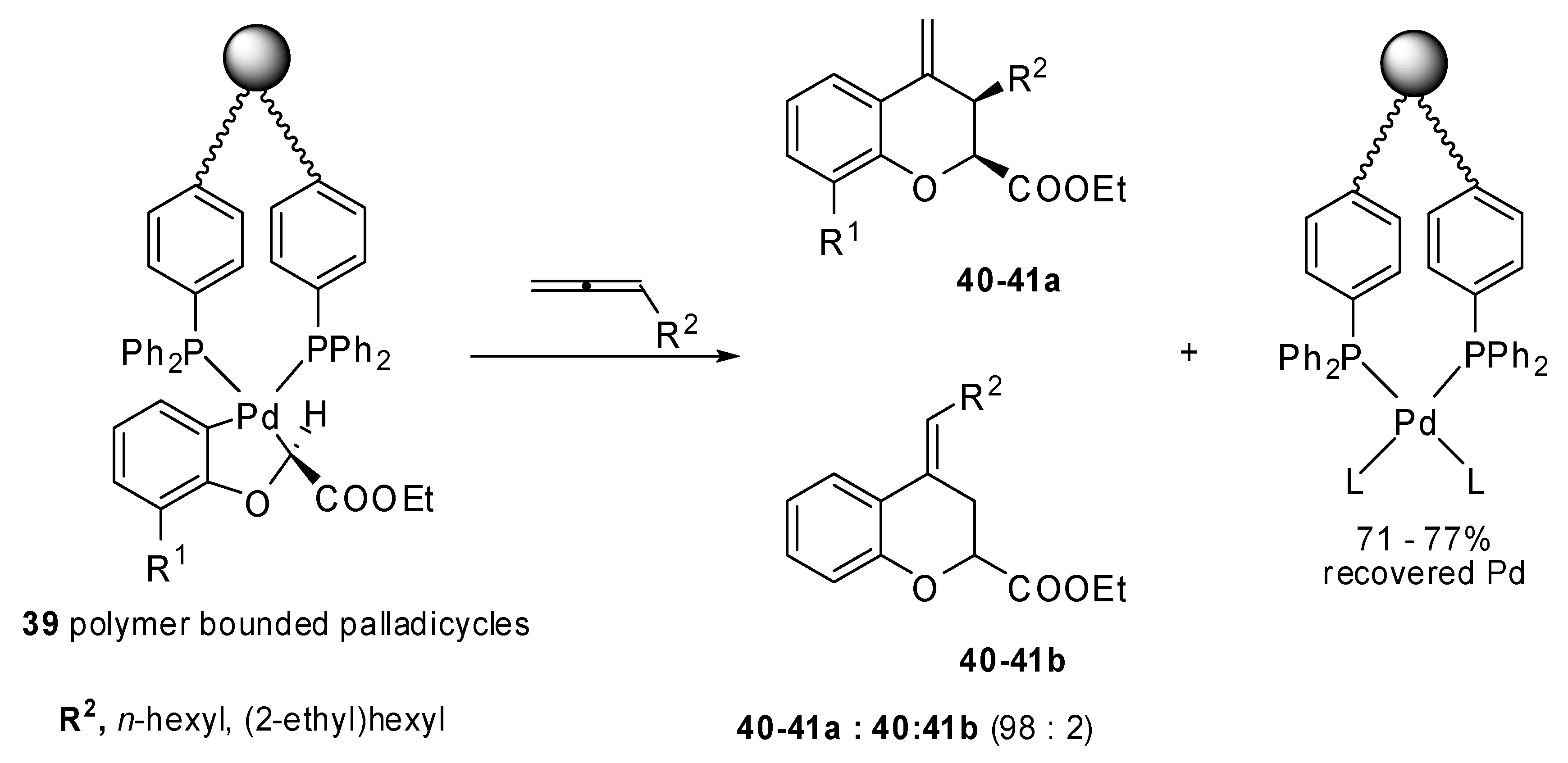
The insertion of monosubstituted allenes into stable oxapalladacycle
38 was studied by Malinakova and coworkers [
26]. The proposed methodology allowed the synthesis of valuable 2,3-disubstituted 3,4-dihydro-2
H-1-benzopyrans (
Scheme 18), which could not be prepared by the classical palladium-catalyzed benzoannulation. Two adjacent stereogenic centers were generated and different, relevant benzopyrans were obtained with potential medicinal properties. The same reaction was applied to the polymer bound palladicycle
39 with remarkable results since more than 70% Pd was recovered (
Scheme 19) [
27].
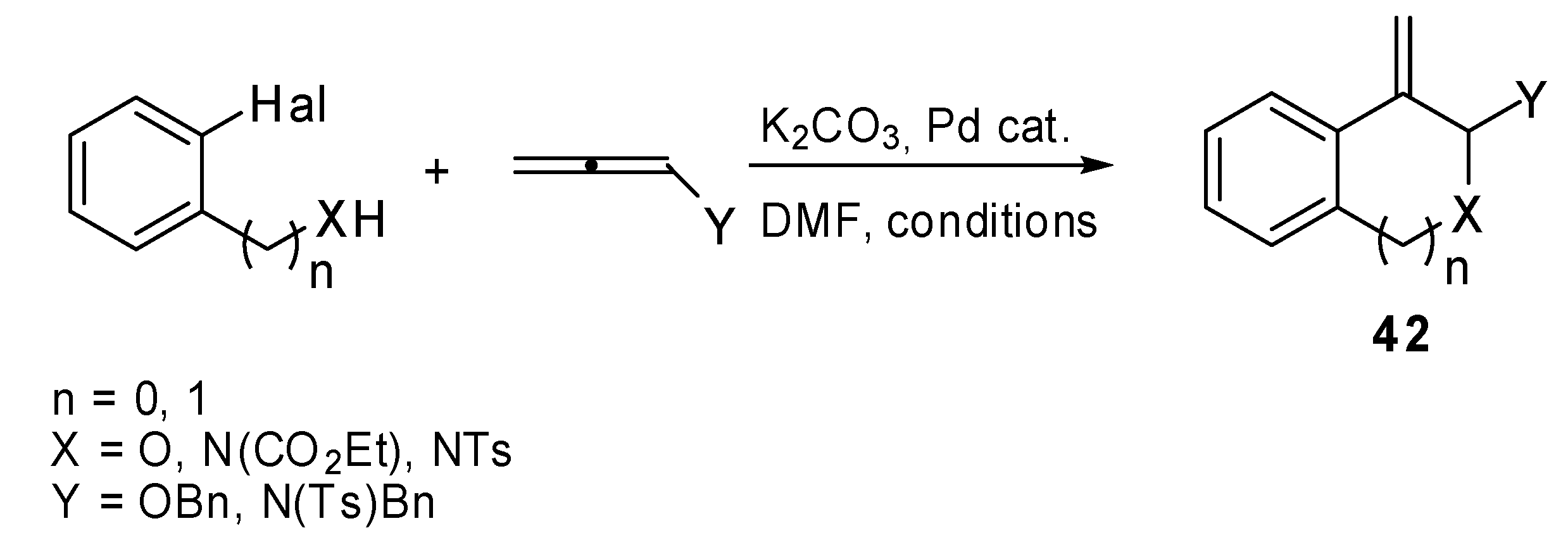
Recently a synthesis of condensed heterocycles
42 was proposed
. It exploited the intramolecular Heck reaction applied to heteroatom substituted allenes and aryl halides (
Scheme 20) [
28]. The initially formed arylpalladium complex added to the allene and produced the corresponding π-allylpalladium intermediate, which readily underwent intramolecular nucleophilic attack by an oxygen or a nitrogen affording the annulated product
42. Despite the presence of two possible attack positions, α or γ, only the α-site attack product was recovered regardless of the bulkiness of the allene substituent. There are two hypotheses to explain this selectivity. The electronegativity of the allene heteroatom renders the α position more electron positive than γ-position or the palladium complex is reductively eliminated with the assistance of the heteroatom and the cyclization proceeds without the participation of the palladium complex.
Scheme 18.
Synthesis of 2,3-disubstituted 3,4-dihydro-2H-1-benzopyrans.
Scheme 18.
Synthesis of 2,3-disubstituted 3,4-dihydro-2H-1-benzopyrans.
Scheme 19.
Synthesis of 2,3-disubstituted 3,4-dihydro-2H-1-benzopyrans using a polymer bounded palladicycle.
Scheme 19.
Synthesis of 2,3-disubstituted 3,4-dihydro-2H-1-benzopyrans using a polymer bounded palladicycle.
Scheme 20.
Annulation reaction of heteroatom-substituted allenes.
Scheme 20.
Annulation reaction of heteroatom-substituted allenes.
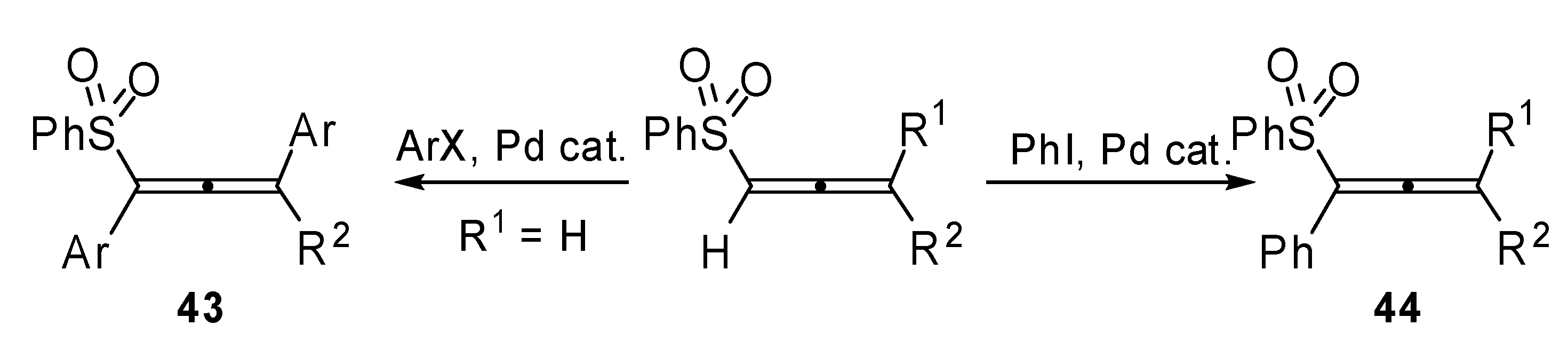
The first example of the Heck type allenylation of aryl halides with allenes was described by Ma and coworkers [
29]. 1,3-Double arylated allenes
43 were recovered when 3-monosubstituted 1,2-allenyl sulfones were used as reagents. Whereas the 1-monoarylation products
44 were obtained in the case of 3,3-disubstituted 1,2-allenyl sulfones (
Scheme 21). The regioselectivity of the intermolecular carbopalladation shown, was completely opposite to what had been previously reported in literature.
Scheme 21.
Heck type allenylation of aryl halides with allenes.
Scheme 21.
Heck type allenylation of aryl halides with allenes.
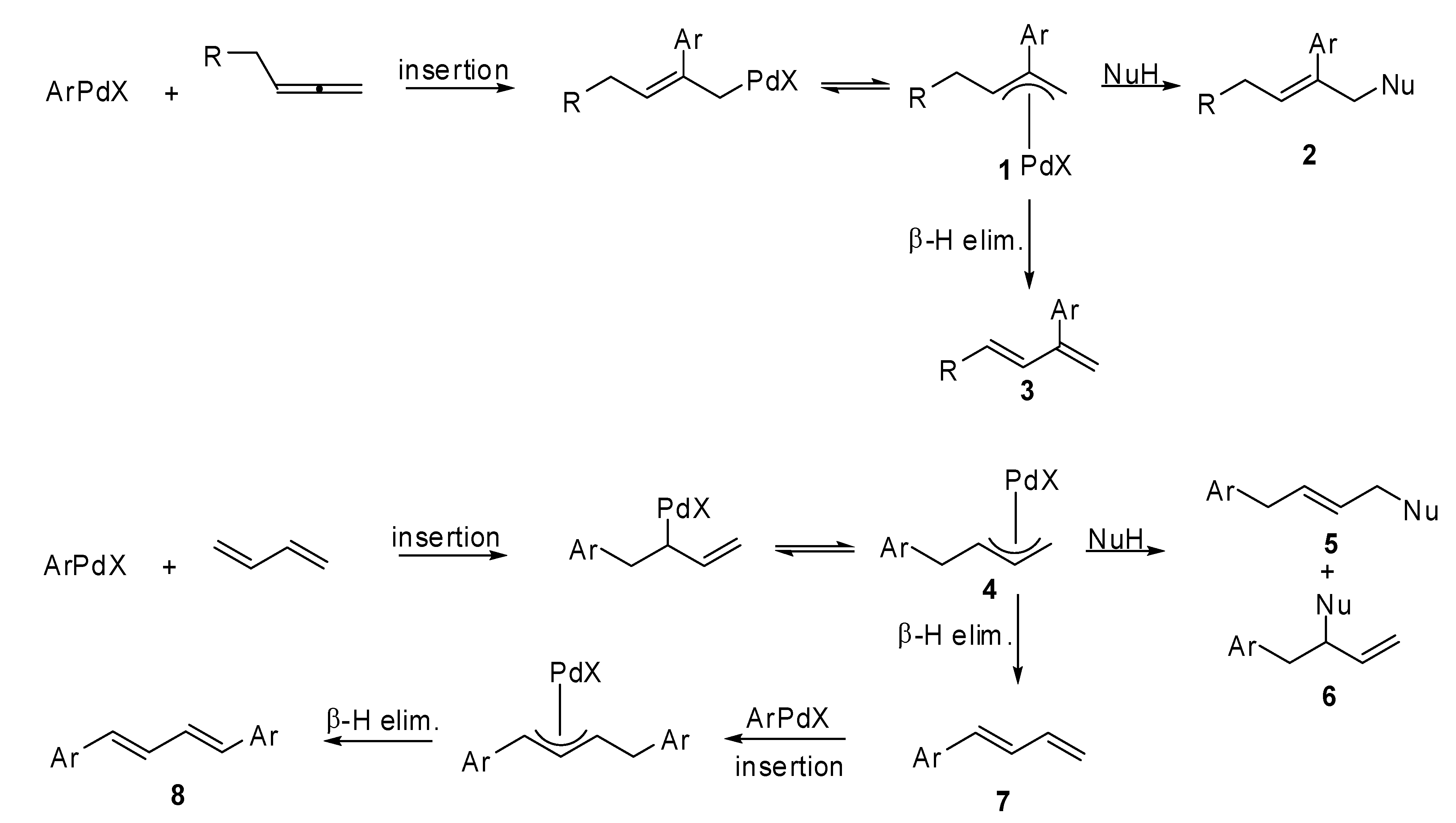
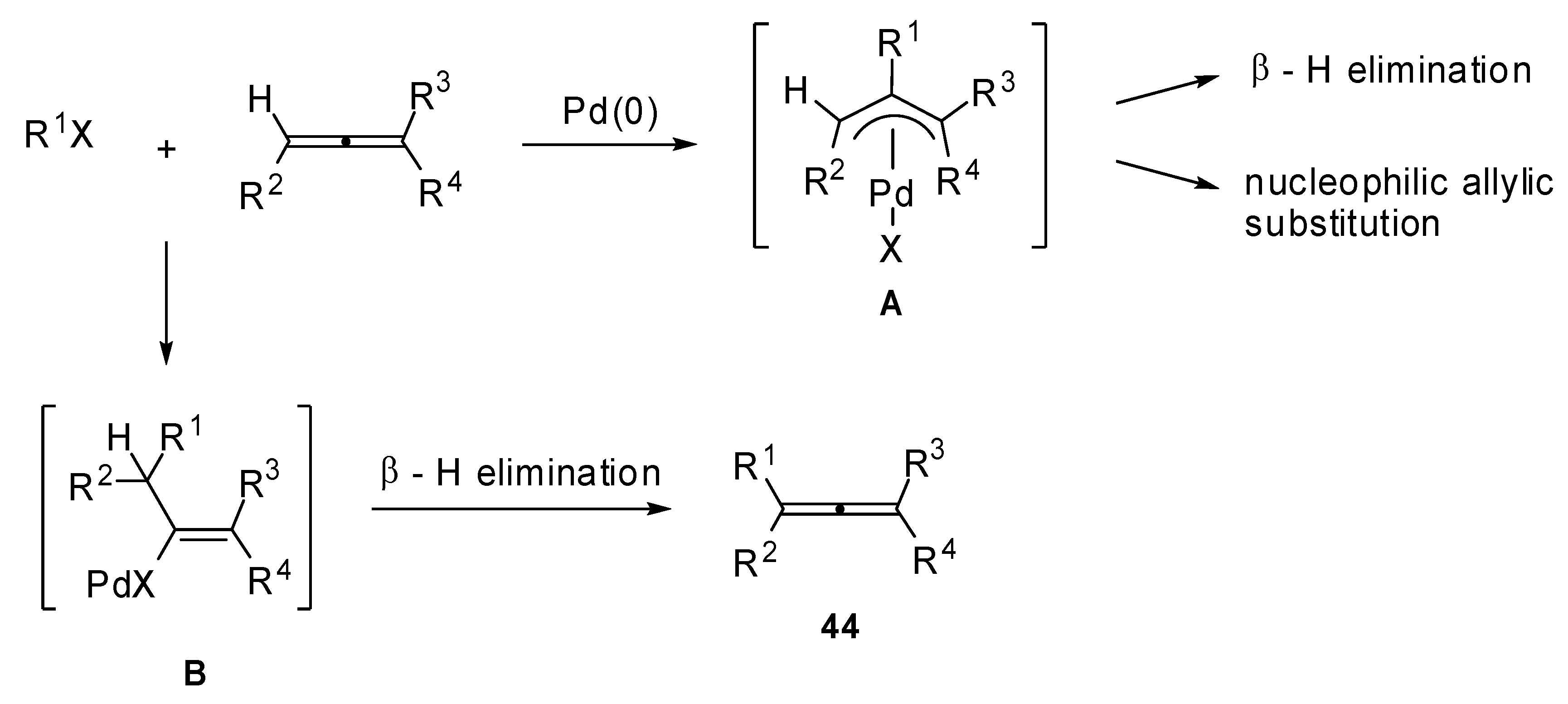
In fact, the carbopalladation of allenes normally affords a π-allylpalladium intermediate
A (
Scheme 22), which may undergo a β-hydride elimination or an allylic substitution affording nonallenic products. The formation of a vinylic palladium intermediate
B is also reported in literature. The subsequent carboxylation leads to the formation of α,β-unsaturated alkenoates. Thus, the regioselectivity of the allene carbopalladation can be determined by the delocalization of the π-allylpalladium intermediate
A and by the high energy/reactivity of the allene that will be formed by the β-H elimination of the vinylic intermediate
B, as described in
Scheme 22.
Scheme 22.
Proposed mechanism for the carbopalladation of allenes.
Scheme 22.
Proposed mechanism for the carbopalladation of allenes.
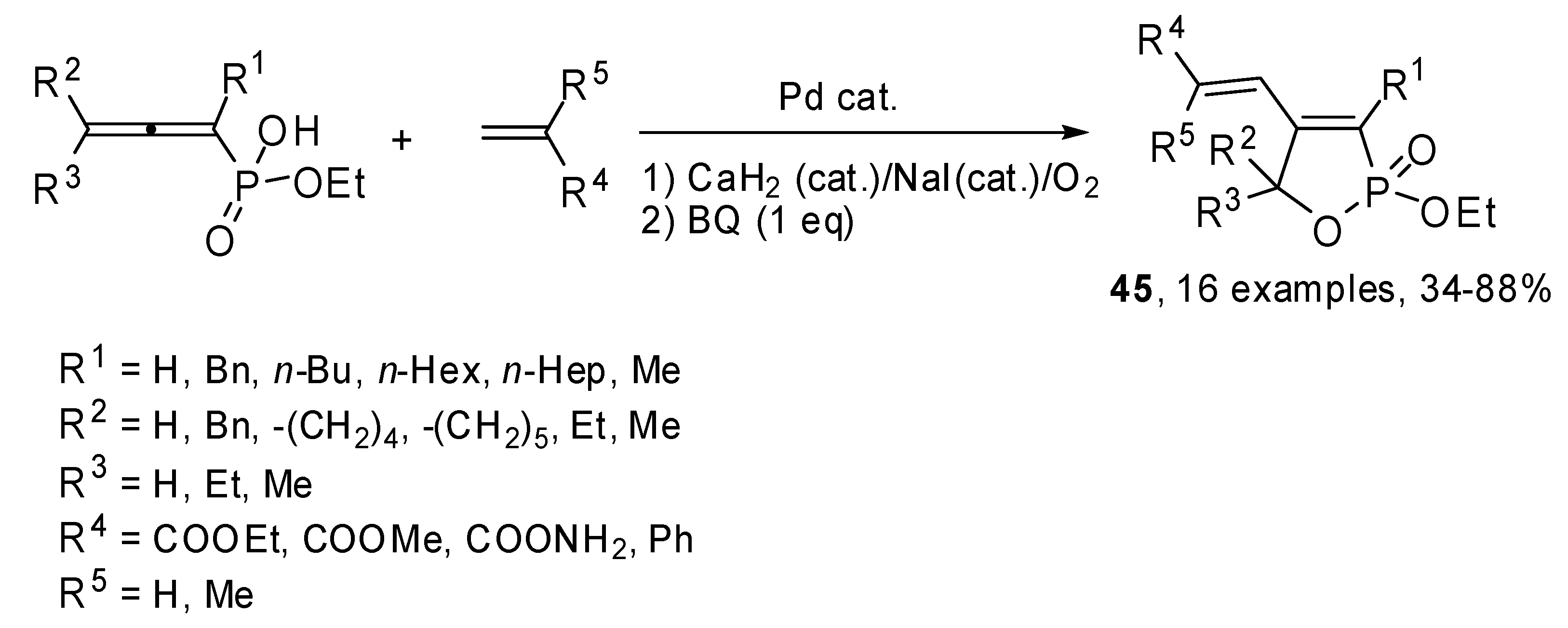
The same authors studied the cyclization−Heck reactions of monoesters of 1,2-allenyl phosphonic acids with allenes. As shown in
Scheme 23, the reaction regio- and stereoselectively afforded 4-(1-
Z-alkenyl)-2-ethoxy-2,5-dihydro[1,2]oxaphosphole 2-oxides
45. Coupling were carried out in oxidative conditions, using CaH
2(cat.)/NaI/O
2 or benzoquinone to regenerate Pd(II) from the
in situ formed Pd(0) [
30].
Scheme 23.
Cyclization-Heck reactions of monoesters of 1,2-allenyl phosphonic acids with allenes.
Scheme 23.
Cyclization-Heck reactions of monoesters of 1,2-allenyl phosphonic acids with allenes.
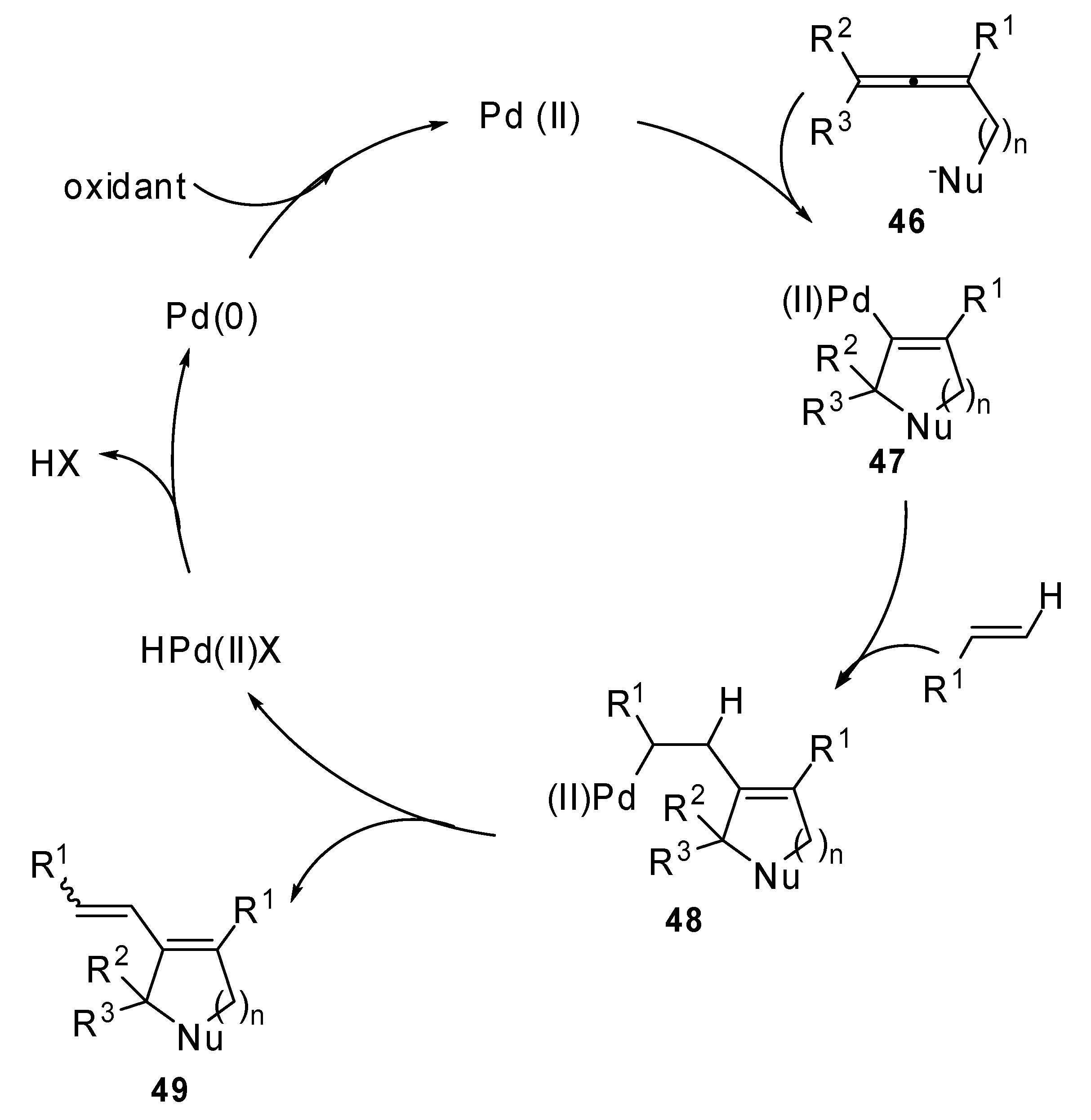
The proposed mechanism is described in
Scheme 24: an
endo-mode cyclic nucleopalladation of allene
46 would form the cyclic palladium intermediate
47. Then, the carbon-carbon alkene double bond is inserted into the C–Pd bond of
47 to form the complex
48. This afforded the final product
49 after a β-H elimination.
Scheme 24.
Proposed catalytic cycle for palladium mediated cyclization-Heck reaction.
Scheme 24.
Proposed catalytic cycle for palladium mediated cyclization-Heck reaction.
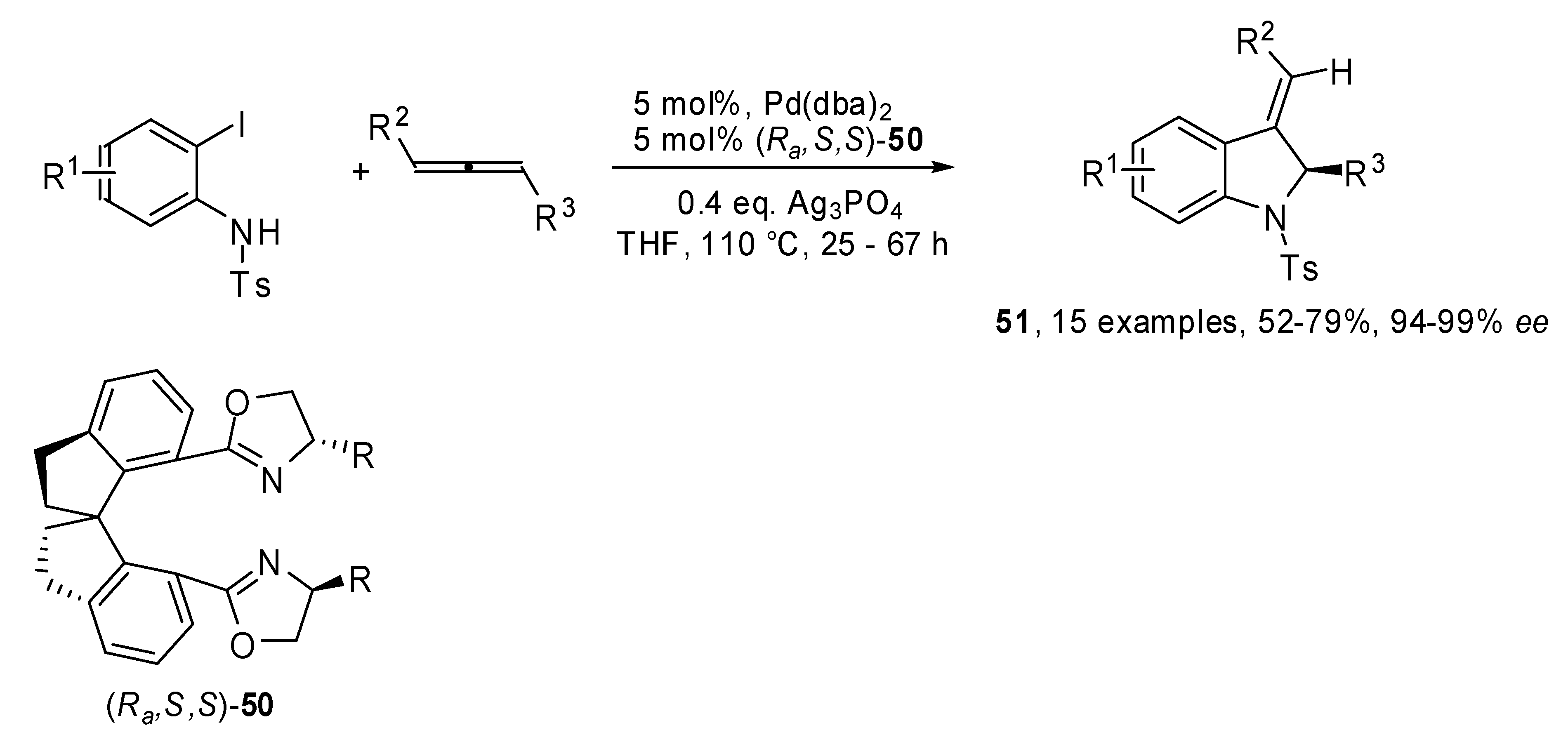
A new chiral spiro-bisoxazoline ligand β-naphtylmethyl-substituted spiro-BOX
50 was developed by the same group and applied to the enantioselective heteroannulations between allenes and 2-iodoanilines [
31]. As shown in
Scheme 25, the coupling produced 3-alkylideneindolines
51 in good yields and remarkable enantiomeric excesses.
Scheme 25.
Enantioselective heteroannulations between allenes and 2-iodoanilines.
Scheme 25.
Enantioselective heteroannulations between allenes and 2-iodoanilines.
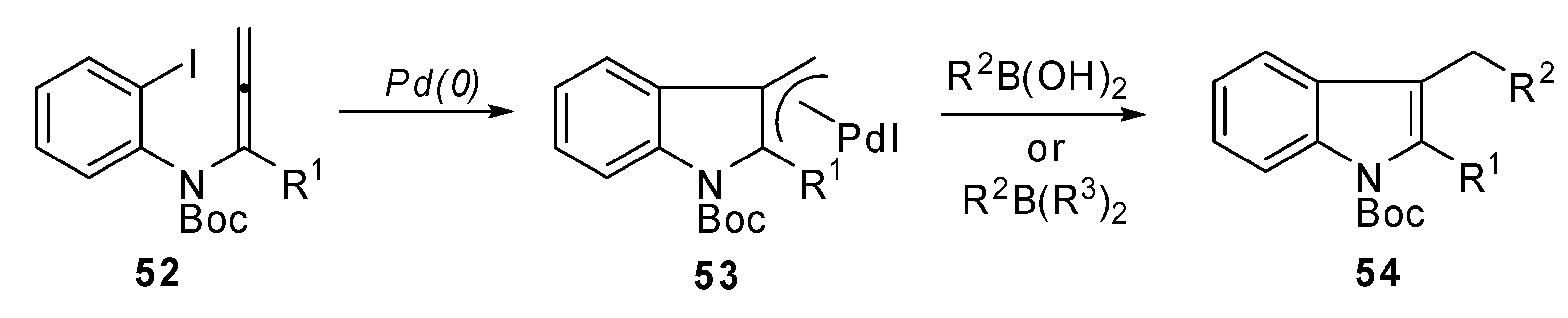
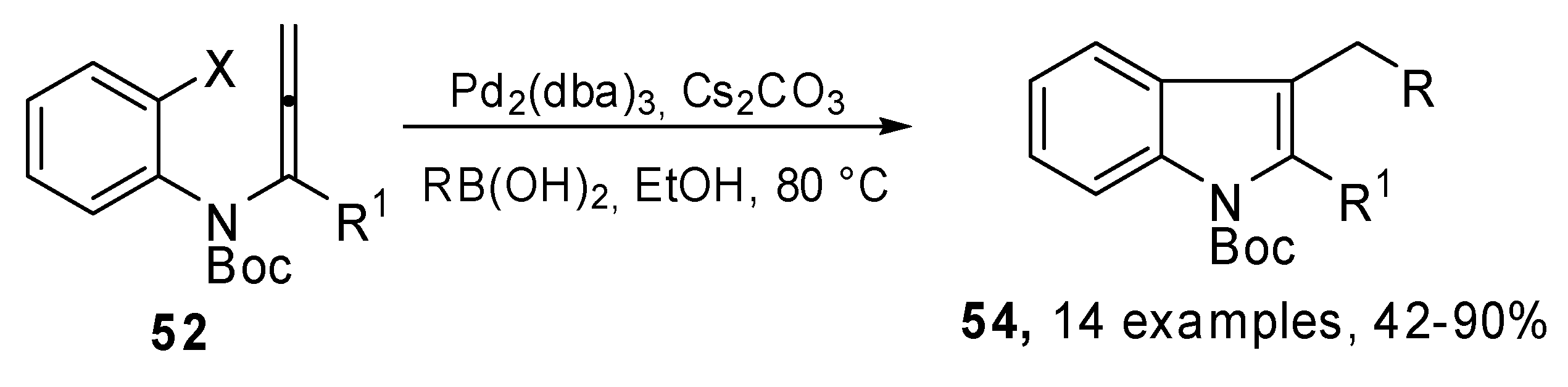
A new synthesis of 2,3-disubstituted indole derivatives 54 was proposed by Sasaki et al. It was based on an intramolecular carbopalladation-anion capture cascade. Several N-(o-halophenyl)-allenamides 52 were used as the starting reagents. Moreover, the introduction of an appropriate silicon group to the α-position of the allenamide, afforded the 2-silyl substituted indole derivatives, which are useful substrates for further Pd(0) catalyzed transformations at the C2 position.
The general strategy, illustrated in
Scheme 26, was based on the utilization of a
N-(
o-halophenyl)allenamide
52 which bore a substituent (R
1) at the α-position of the allenamide. In this case a π-allylpalladium intermediate
53 could be generated
via carbopalladation and could be trapped by a suitable nucleophile, such as an aryl or alkenyl boronic acid or alkylborane (
Scheme 27) [
32].
Scheme 26.
General strategy for the intramolecular carbopalladation-anion capture cascade of N-(o-halophenyl)allenamides.
Scheme 26.
General strategy for the intramolecular carbopalladation-anion capture cascade of N-(o-halophenyl)allenamides.
Scheme 27.
Synthesis of 3-substituted and 2,3- disubstituted indoles.
Scheme 27.
Synthesis of 3-substituted and 2,3- disubstituted indoles.
The methodology was then extended to the preparation of indoles-2,3-quinodimethanes which are highly reactive dienes that readily undergo Diels–Alder cycloaddition to get tetrahydrocarbazoles and related compounds [
33].
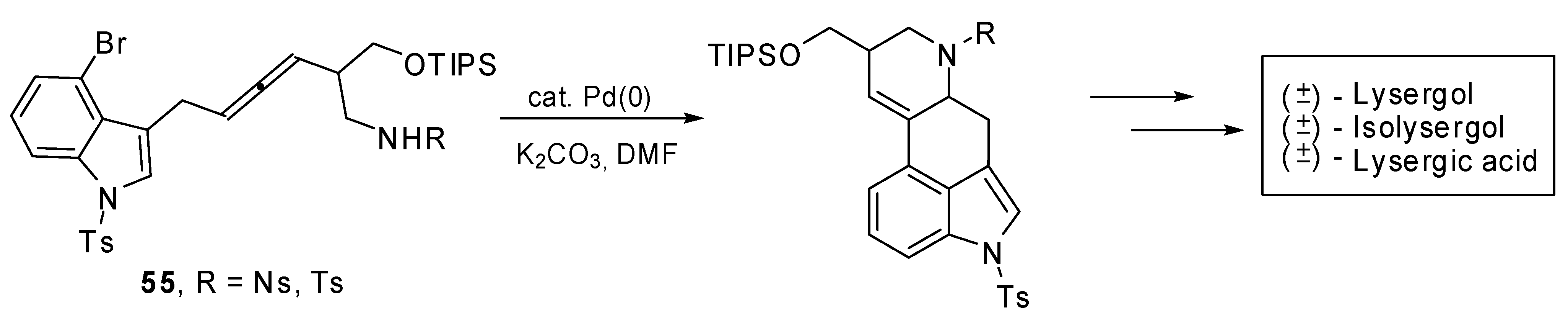
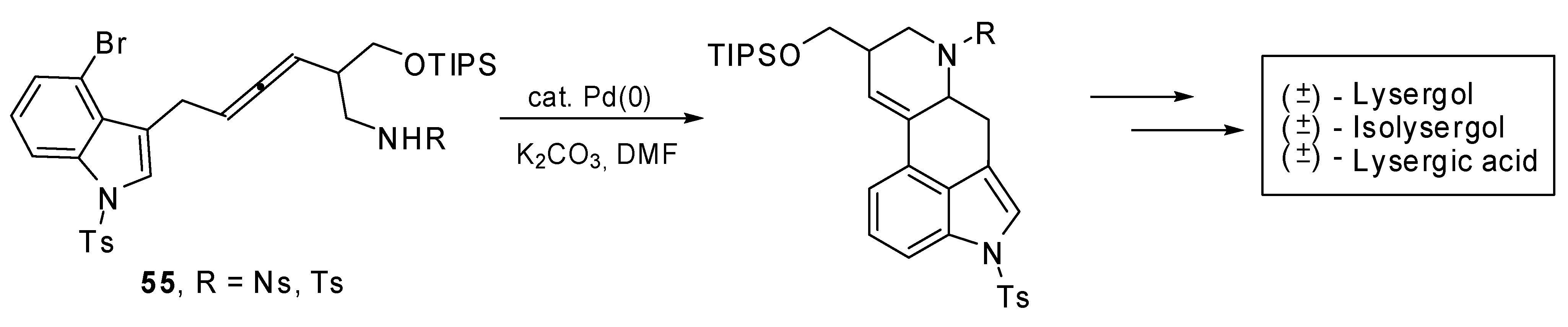
The palladium catalyzed domino cyclization of amino allenes
55 was exploited to build the C/D ring system of ergot alkaloids. The total syntheses of (±)-lysergic acid, (±)-lysergol and (±)-isolysergol were achieved (
Scheme 28) with this bisannulation as the key step [
34].
Scheme 28.
Synthesis of (±)-lysergic acid, (±)-lysergol and (±)-isolysergol by palladium catalyzed domino cyclization of amino allenes.
Scheme 28.
Synthesis of (±)-lysergic acid, (±)-lysergol and (±)-isolysergol by palladium catalyzed domino cyclization of amino allenes.
The best reaction results in terms of yields and diastereoselectivity were obtained using Pd(PPh3)4 as the catalyst, K2CO3 as the base in DMF at 100 °C.
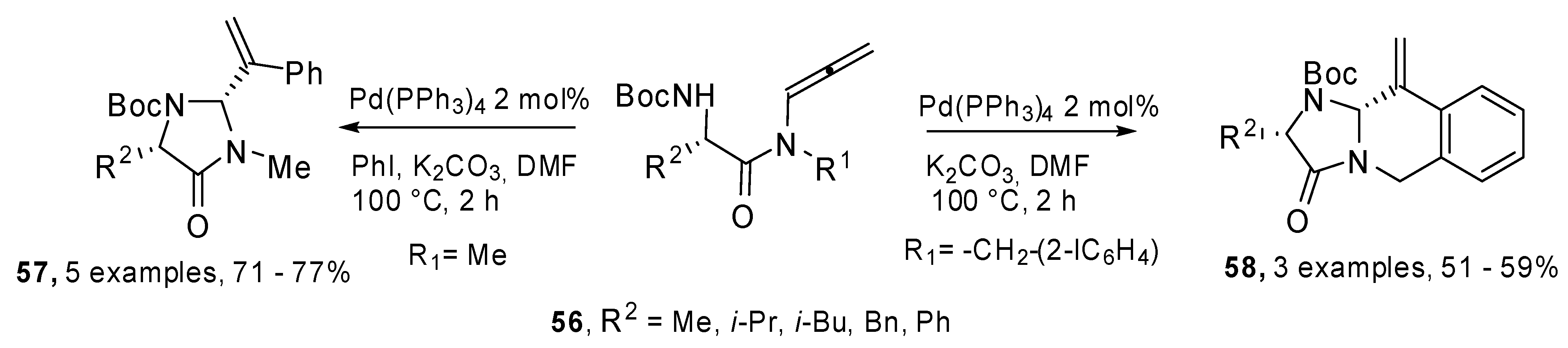
Allenamides of α-amino acids
56 were used for the preparation of enantiopure imidazolin-4-ones
57 and imidazoisoquinolinones
58 by means of a domino carbopalladation/allylic amination process. The authors also demonstrated the feasibility of the heterocyclization process with an amide group in the tether, without any interference of the carbonyl group (
Scheme 29) [
35].
Scheme 29.
Heterocyclization reactions of allenamides by carbopalladation-intramolecular amination.
Scheme 29.
Heterocyclization reactions of allenamides by carbopalladation-intramolecular amination.
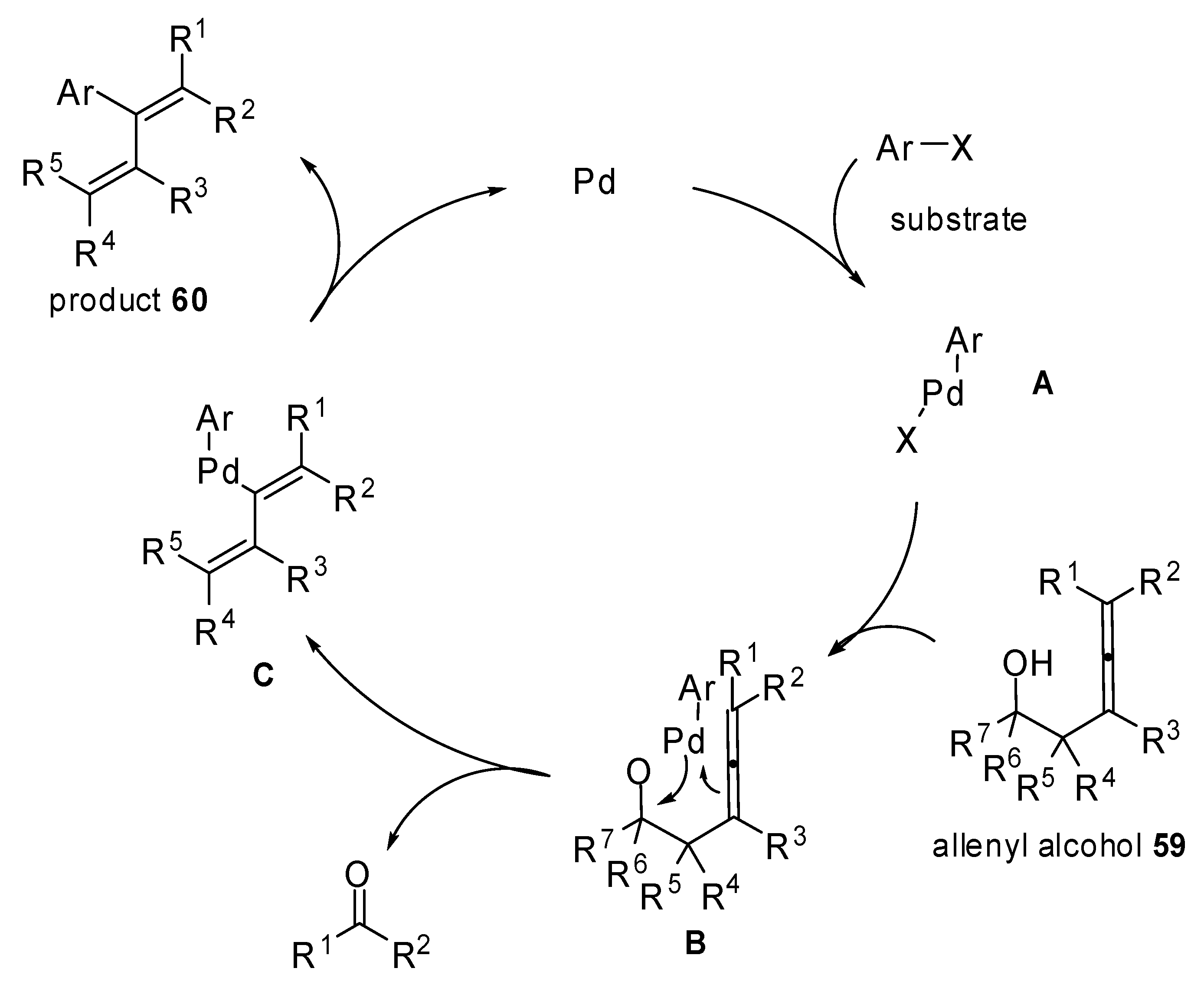
A new method for the synthesis of 2-aryl-1,3-alkadienes
60 is illustrated in
Scheme 30. Palladium-catalyzed 1-methylene-2-propenylation reactions of aryl bromides with 3,4-alkadien-1-ols
59 were exploited and a palladium mediated retro-allylation allowed the alcohols
59 to act as 1-methylene-2-propenyl metals. Moreover, they are inert in air and readily available [
36].
Scheme 30.
Synthesis of 2-aryl-1,3-alkadienes by aryl bromides and 3,4-alkadien-1-ols.
Scheme 30.
Synthesis of 2-aryl-1,3-alkadienes by aryl bromides and 3,4-alkadien-1-ols.
The mechanism hypothesized by the authors is reported in
Scheme 31. The palladium alcoxide
B is formed after the oxidative addition and the ligand exchange. Then a C–C bond cleavage proceeds via a six-membered cyclic transition state selectively providing the intermediate
C. Finally the reductive elimination will furnish the desired 2-aryl-1,3-alkadienes
60.
Scheme 31.
Hypothesized mechanism for the achievement of 2-aryl-1,3-alkadienes.
Scheme 31.
Hypothesized mechanism for the achievement of 2-aryl-1,3-alkadienes.
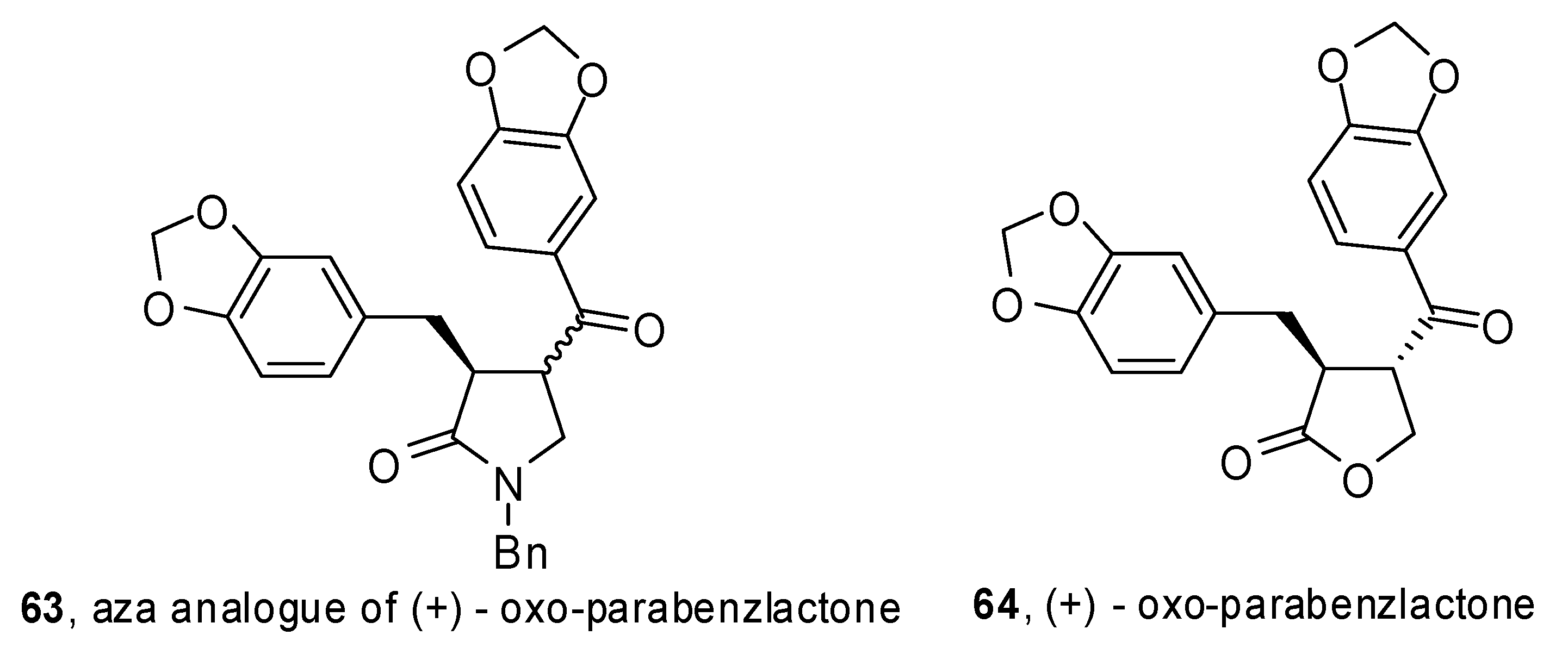
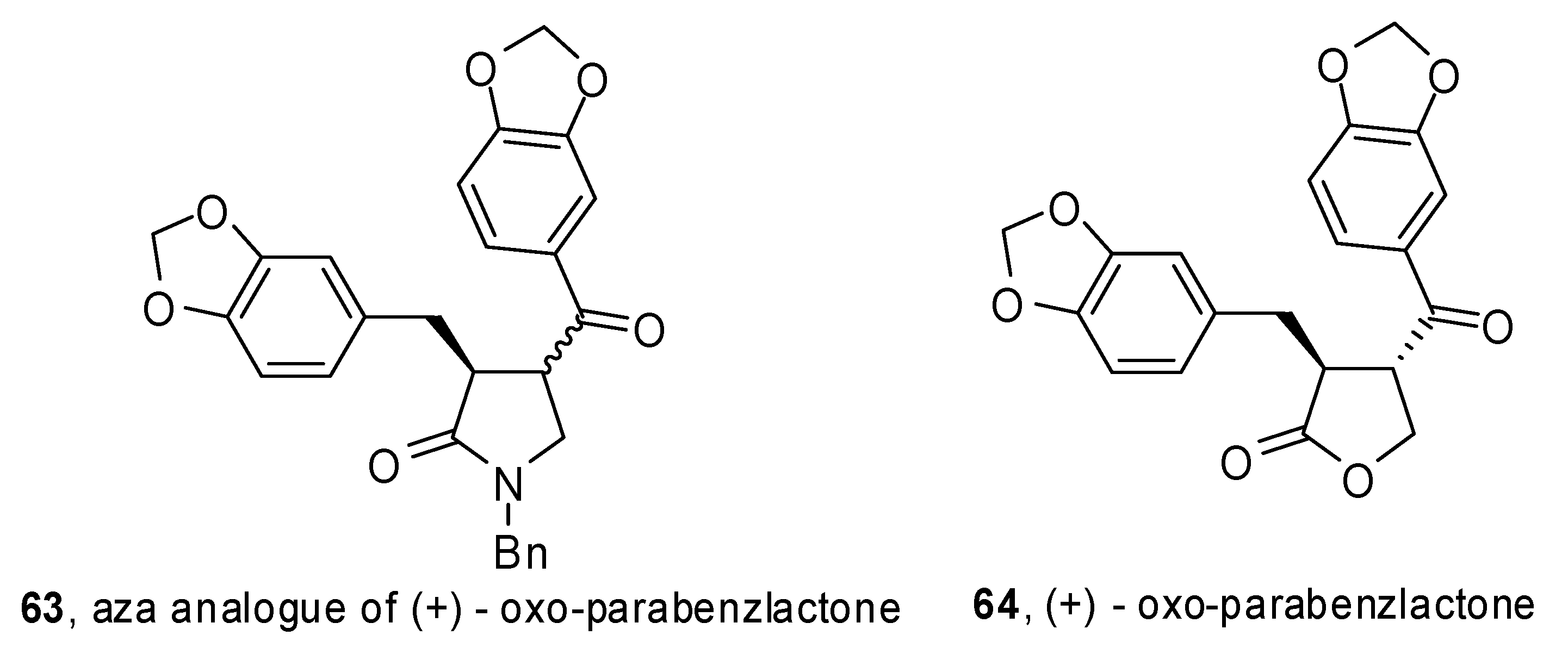
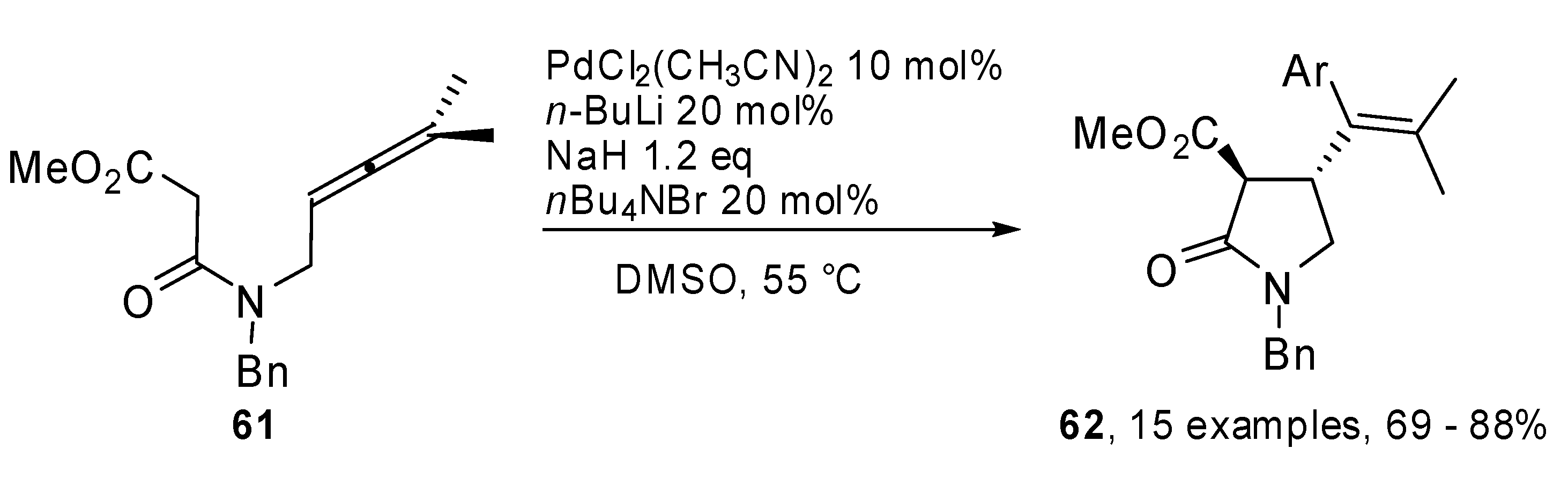
A phosphine-free Pd-catalyzed allene carbopalladation/allylic alkylation sequence was exploited by Poli and coworkers to get 4-(α-styryl) γ-lactams
62 from β-aminoallene
61 (
Scheme 32) [
37]. High yields were obtained for electron-rich as well as electron-poor aryl iodides. Furthermore the reaction was completely regio- and stereoselective
versus the 5-
exo trans product. The synthetic sequence was extended to the preparation of an aza analogue of (+) oxo-parabenzlactone (
63,
Figure 1), a naturally occurring lignin.
Scheme 32.
Synthesis of 4-(α-styryl) γ-lactams by β-aminoallene.
Scheme 32.
Synthesis of 4-(α-styryl) γ-lactams by β-aminoallene.
Figure 1.
Aza analogue of oxo-parabenzlactone.
Figure 1.
Aza analogue of oxo-parabenzlactone.
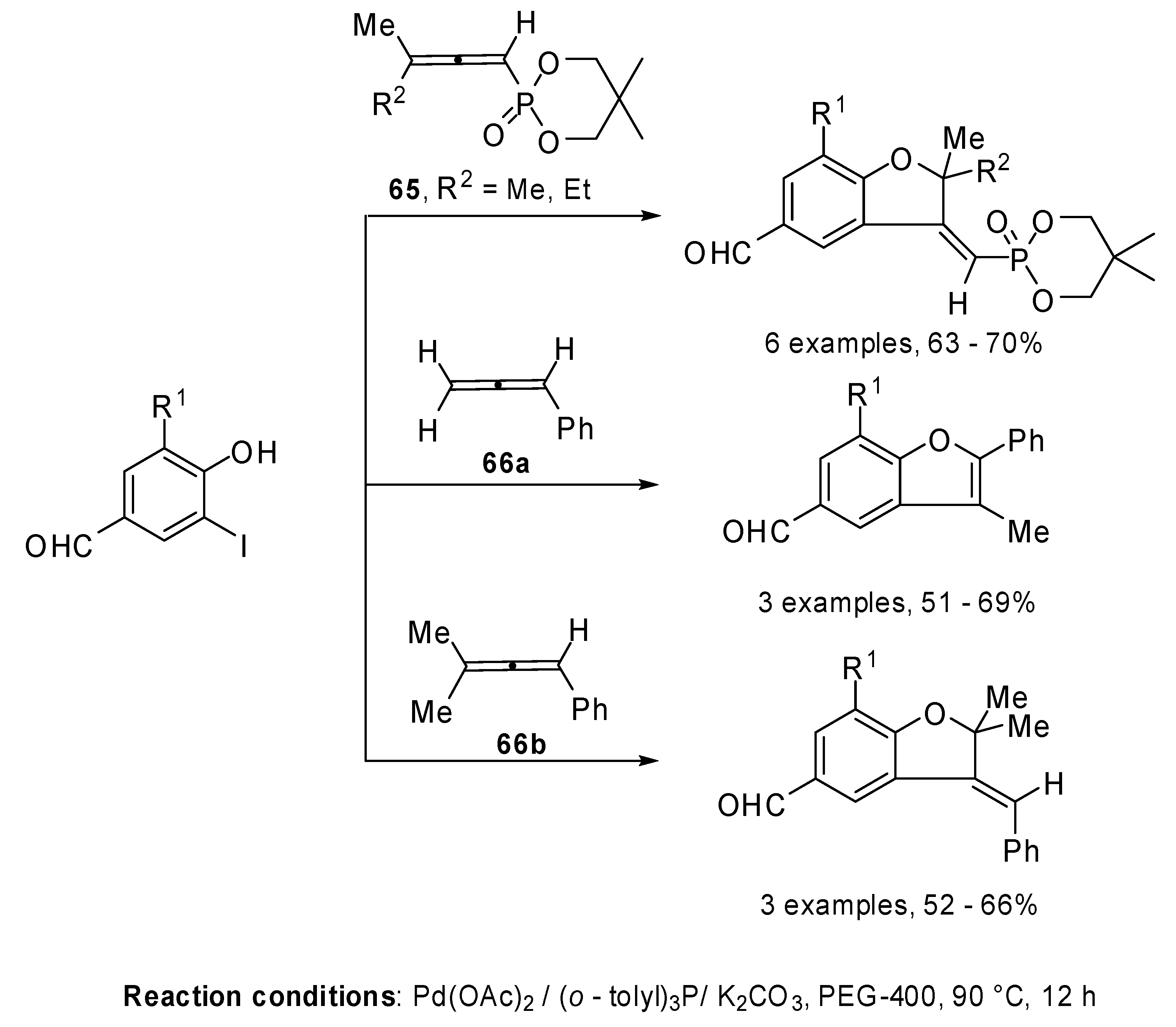
The regioselective palladium catalyzed coupling reaction of allenylphosphonates
65 and phenylallenes
66 with functionalized iodophenols, 2-iodobenzoic acid and 2-iodobenzyl alcohol was investigated [
38]. Recently a similar reactivity stydy using PEG 400 as the solvent was described. The reaction allowed the regioselective formation of aldehyde-functionalized benzofuranes and benzopyrans. The above mentioned results demonstrated that a [β,γ] attack on the allene was preferred when benzofurans or benzopyrans were formed, except in the case of PhC=C=CCH
2 (
66a) where a [β,α] attack was observed (
Scheme 33) [
39].
Scheme 33.
Reactions of allenylphosphonates and phenylallenes with functionalized iodophenols, 2-iodobenzoic acid in PEG 400.
Scheme 33.
Reactions of allenylphosphonates and phenylallenes with functionalized iodophenols, 2-iodobenzoic acid in PEG 400.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
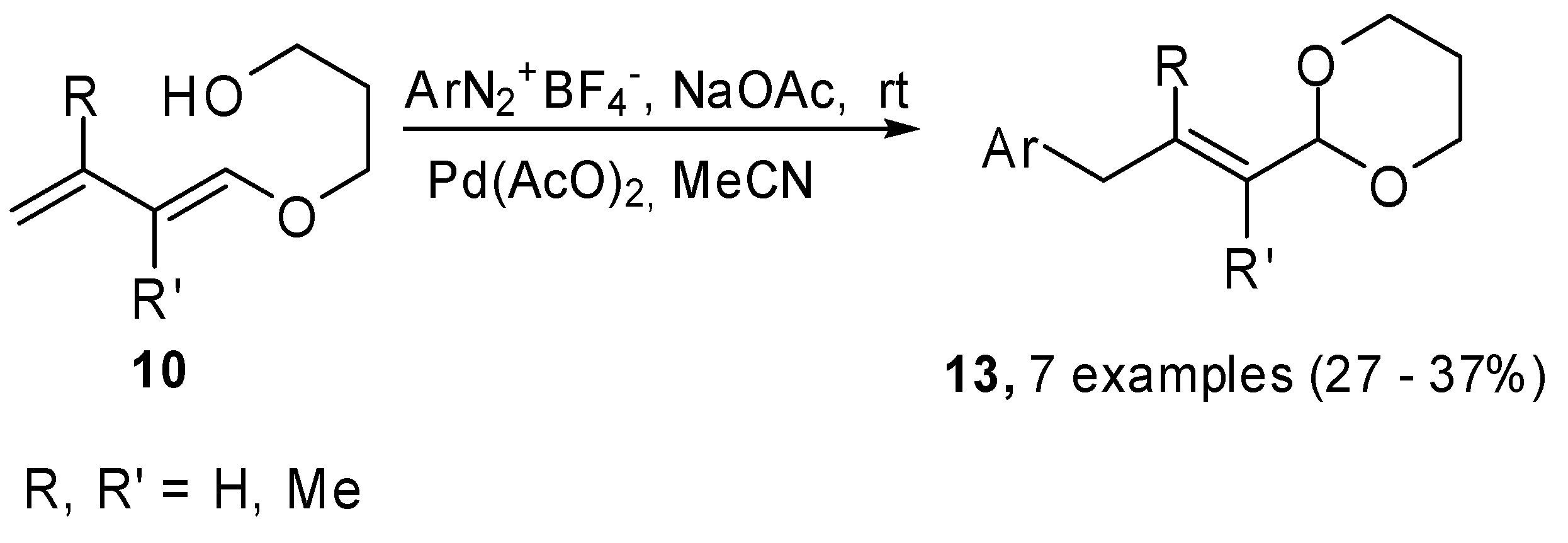{kind=link}
{kind=link}
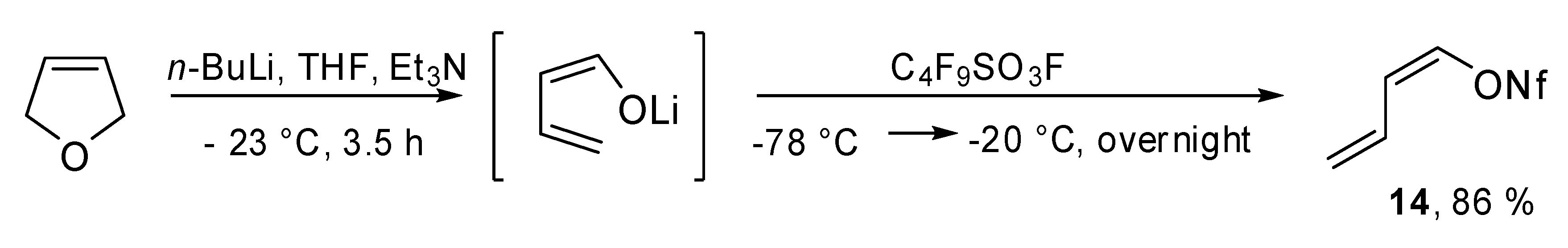{kind=link}
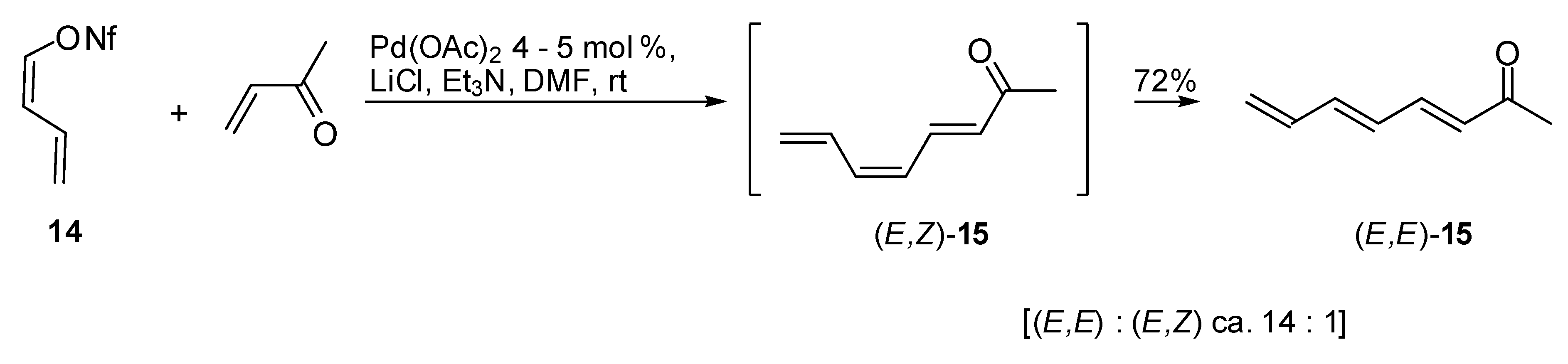{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}