Access to Original Vinylic Chlorides in the Quinazoline Series via a Monoelectronic Transfer Reaction Approach


Abstract
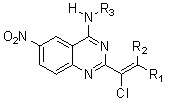
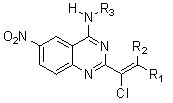
:
1. Introduction
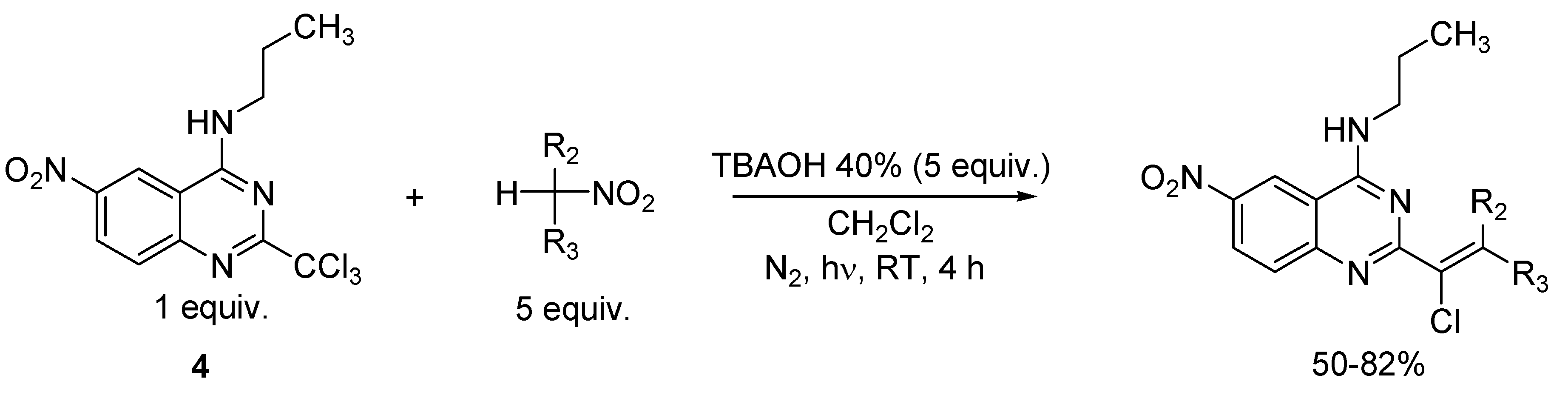
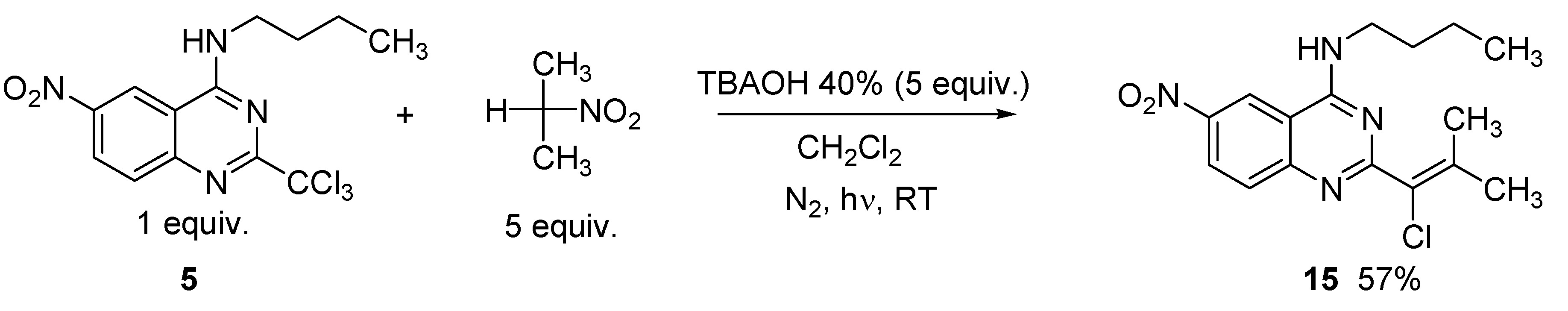
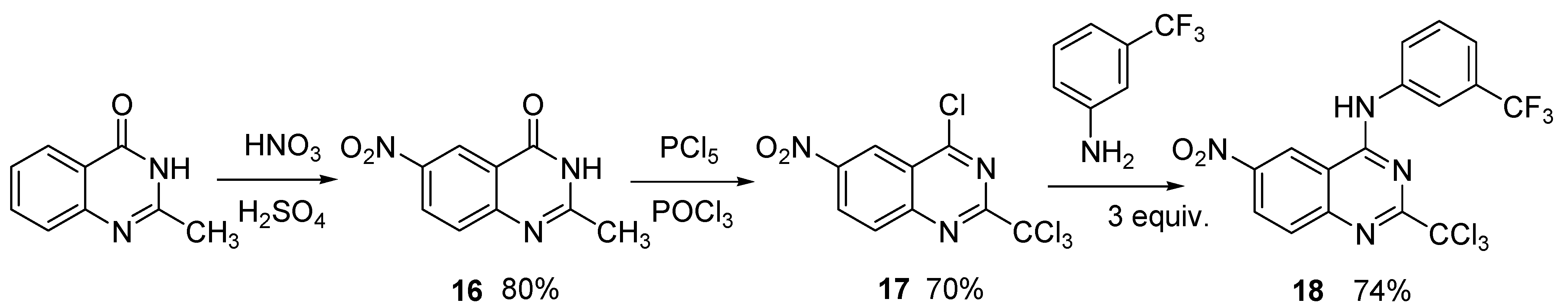
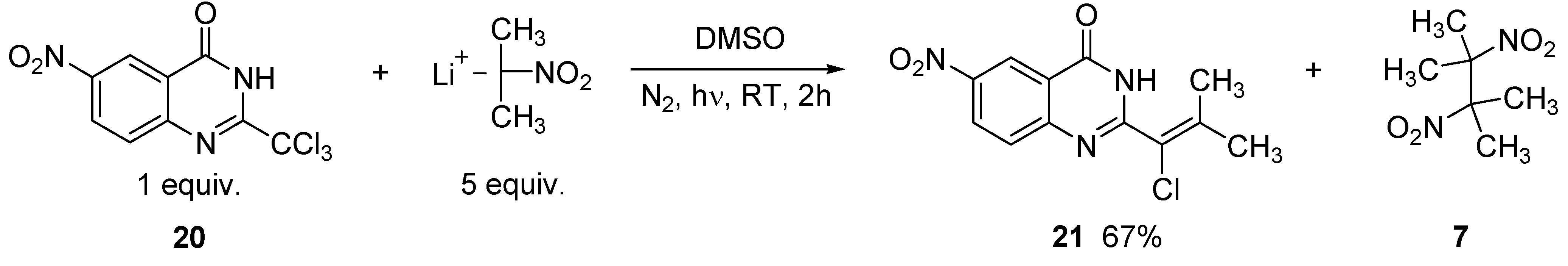
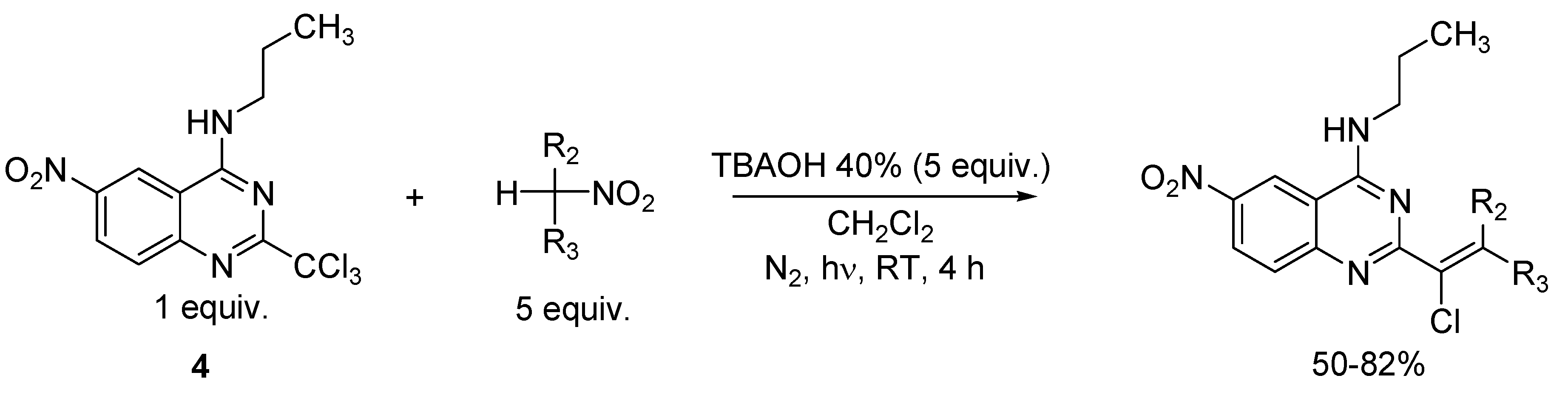
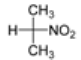
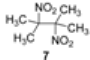
2. Results and Discussion



{kind=link}
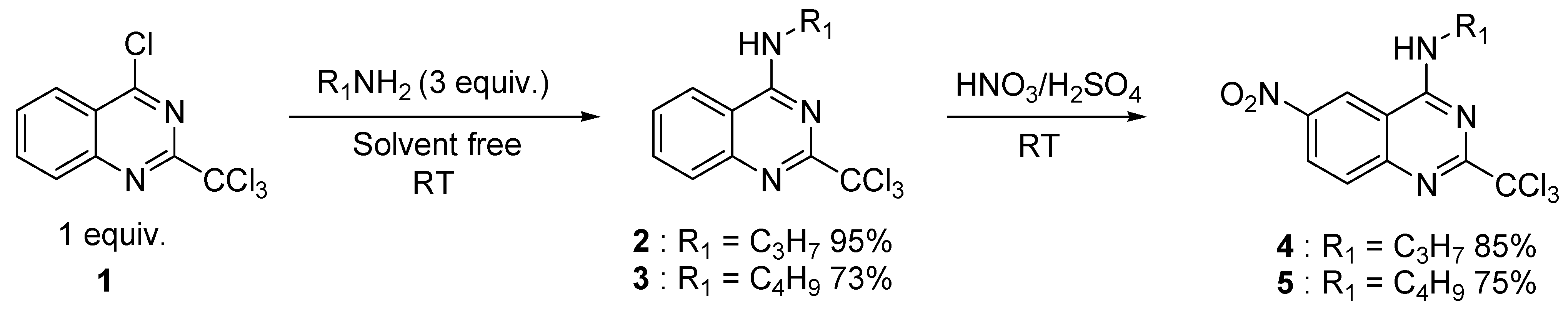{kind=link}
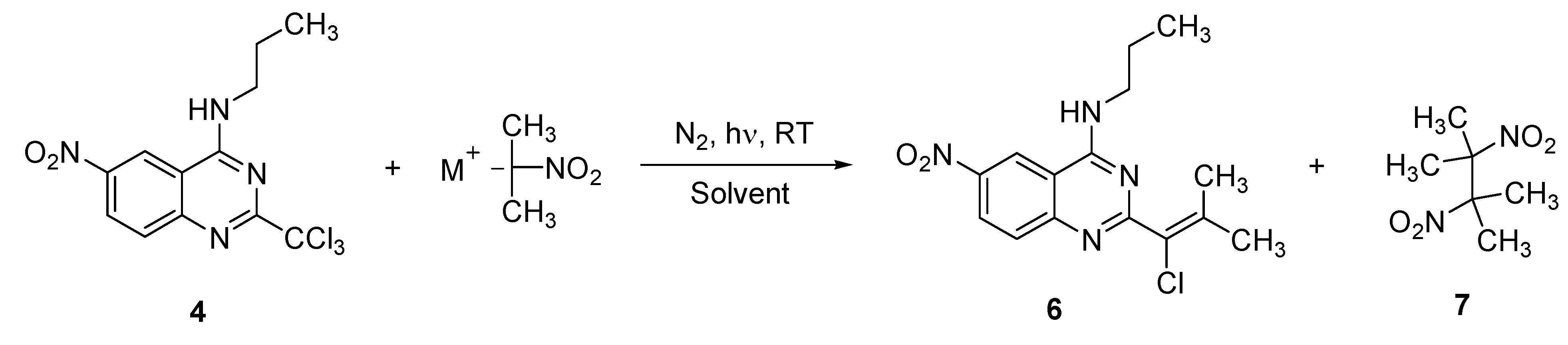{kind=link}
{kind=link}
{kind=link}
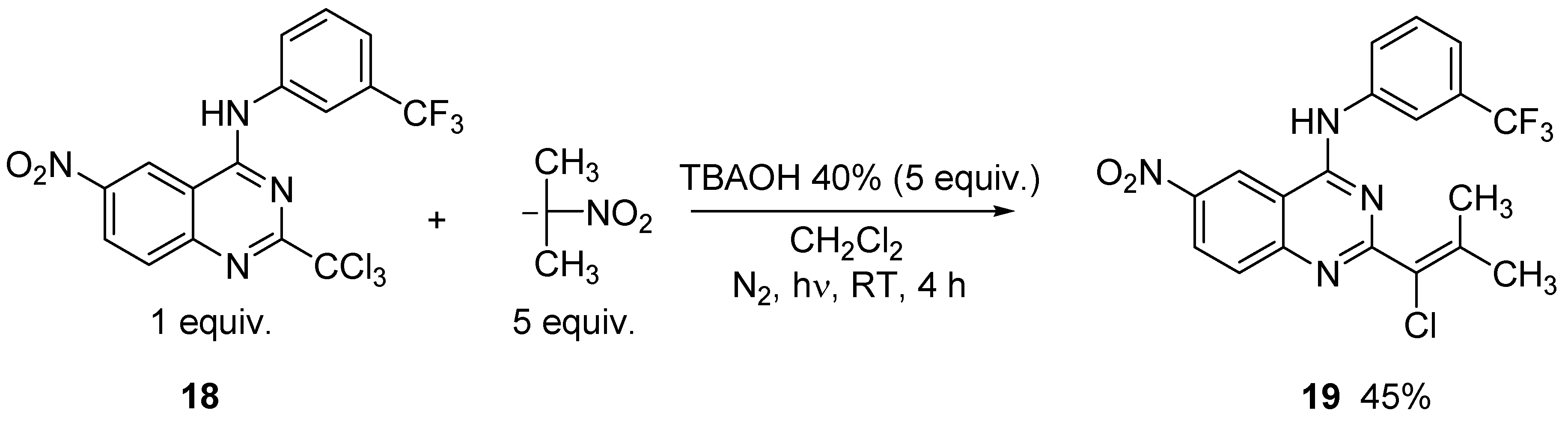{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run* | M+ | Solvent | 2-Nitropropane (equiv.) | Inhibitor | Yield of 6 (%) |
|---|---|---|---|---|---|
| 1 | NBu4 | CH2Cl2H2O | 2 | - | 60 |
| 2 | NBu4 | CH2Cl2H2O | 4 | - | 75 |
| 3 | NBu4 | CH2Cl2H2O | 5 | - | 82 |
| 4 | NBu4 | CH2Cl2H2O | 6 | - | 83 |
| 5 | Li | DMF | 5 | - | 53 |
| 6 | Li | DMSO | 5 | - | 70 |
| 7 | Li | DMSO | 5 | CuCl2 (1 equiv.) | 25 |
| 8 | NBu4 | CH2Cl2H2O | 5 | TEMPO (1 equiv.) | 0 |
| 9 | NBu4 | CH2Cl2H2O | 5 | O2 bubbling | 0 |
| 10 | NBu4 | CH2Cl2H2O | 5 | p-dinitrobenzene | 35 |
| Nitroalkane | Vinylic chloride | Dimer | Yield of vinylic chloride (%) |
|---|---|---|---|
 |  |  | 82 |
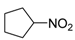 | 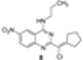 |  | 60 |
 | 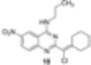 | 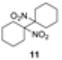 | 66 |
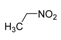 | 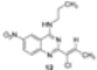 | Not isolated | 56 |
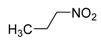 | 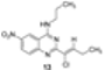 | Not isolated | 50 |
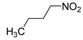 | 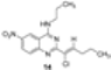 | Not isolated | 52 |





3. Experimental
3.1. General
3.2. Preparation of N-propyl-2-trichloromethylquinazolin-4-amine (2)
3.3. General Procedure for the Preparation of Compounds 4 and 5
3.4. General Procedure for the Synthesis of Vinylic Chlorides 6, 8, 10, 12, 13, 14, 15 and 19
3.5. Preparation of 6-nitro-2-trichloromethyl-N-[(3-trifluoromethyl)]phenylquinazolin-4-amine (18)
3.6. Preparation of 2-(1-chloro-2-methylprop-1-enyl)-6-nitroquinazolin-4(3H)-one (21)
4. Conclusions
Acknowledgements
- Samples Availability: Samples of the compounds are available from the authors.
References
- Verhaeghe, P.; Azas, N.; Gasquet, M.; Hutter, S.; Ducros, C.; Laget, M.; Rault, S.; Rathelot, P.; Vanelle, P. Synthesis and antiplasmodial activity of new 4-aryl-2-trichloromethylquinazolines. Bioorg. Med. Chem. Lett. 2008, 18, 396–401. [Google Scholar]
- Verhaeghe, P.; Azas, N.; Hutter, S.; Castera-Ducros, C.; Laget, M.; Dumètre, A.; Gasquet, M.; Reboul, J.P.; Rault, S.; Rathelot, P.; Vanelle, P. Synthesis and in vitro antiplasmodial evaluation of 4-anilino-2-trichloromethylquinazolines. Bioorg. Med. Chem. 2009, 17, 4313–4322. [Google Scholar] [CrossRef]
- Kabri, Y.; Azas, N.; Dumetre, A.; Hutter, S.; Laget, M.; Verhaeghe, P.; Gellis, A.; Vanelle, P. Original quinazoline derivatives displaying antiplasmodial properties. Eur. J. Med. Chem. 2010, 45, 616–622. [Google Scholar] [CrossRef]
- Ryabukhin, S.V.; Plaskon, A.S.; Volochnyuk, D.M.; Tolmachev, A.A. Chlorotrimethylsilane-mediated synthesis of 2-aryl-1-chloro-1-heteroarylalkenes. Synthesis 2007, 20, 3163–3170. [Google Scholar]
- Vanelle, P.; Rathelot, P.; Maldonado, J.; Crozet, M.P. Consecutive SRN1 and ERC1 reactions in 5-nitroisoquinolines. Tetrahedron Lett. 1994, 35, 8385–8388. [Google Scholar] [CrossRef]
- Vanelle, P.; Benakli, K.; Giraud, L.; Crozet, M.P. Synthesis of tetrasubstituted ethylenic compounds from a gem-trichloro imidazole derivative by electron-transfer reactions. Synlett 1999, 6, 801–803. [Google Scholar]
- Verhaeghe, P.; Rathelot, P.; Rault, S.; Vanelle, P. Convenient preparation of original vinylic chlorides with antiparasitic potential in quinoline series. Lett. Org. Chem. 2006, 3, 891–897. [Google Scholar] [CrossRef]
- Kabri, Y.; Gellis, A.; Vanelle, P. Microwave-assisted synthesis in aqueous medium of new quinazoline derivatives as anticancer precursors. Green Chem. 2009, 11, 201–208. [Google Scholar] [CrossRef]
- Verhaeghe, P.; Rathelot, P.; Gellis, A.; Rault, S.; Vanelle, P. Highly efficient microwave assisted α-trichlorination reaction of α-methylated nitrogen containing heterocycles. Tetrahedron 2006, 62, 8173–8176. [Google Scholar] [CrossRef]
- Kerber, R.C.; Urry, G.W.; Kornblum, N. Radical anions as intermediates in substitution reactions. Carbon alkylation of nitroparaffin salts. J. Am. Chem. Soc. 1964, 86, 3904. [Google Scholar] [CrossRef]
- Burt, B.; Freeman, D.J.; Gray, P.G.; Norris, R.K.; Randles, D. Radical and ionic reactions of tetrabutylammonium aci-nitronates. Tetrahedron Lett. 1977, 35, 3063–3066. [Google Scholar]
- Dehoff, L.H. Nitro- and chloro-derivatives of β-methyl-γ-oxyquinazoline (anhydroacetyl-orthamidobenzamide). J. Prakt. Chem. 1890, 42, 346–360. [Google Scholar] [CrossRef]
- Wan, Z.K.; Wacharasindhu, S.; Levins, C.G.; Lin, M.; Tabei, K.; Mansour, T.S. The scope and mechanism of phosphonium-mediated SNAr reactions in heterocyclic amides and ureas. J. Org. Chem. 2007, 72, 10194–10210. [Google Scholar] [CrossRef]
- Girdler, D.J.; Norris, R.K. Unusual reaction of benzylidine diacetates. Ionic chain mechanism for the substitution of an acetate group by the 2-nitro-2-propanide ion. Tetrahedron lett. 1975, 7, 431–434. [Google Scholar]
- Ono, N.; Miyake, H.; Tamura, R.; Hamamoto, I.; Kaji, A. Free radical chain elimination reaction (ERC1). Conversion of vicinal dinitro compounds or β-nitro sulfones to olefins with tributyltin hybride. Chem. Lett. 1981, 10, 1139–1142. [Google Scholar]
- Kornblum, N.; Boyd, S.D.; Pinnick, H.W.; Smith, R.G. New synthesis of olefins. J. Am. Chem. Soc. 1971, 93, 4316–4318. [Google Scholar]
- Bogert, M.T.; Geiger, G.A. Researches on quinazolines (thirtieth paper). Study of the bromination and nitration of 4-quinazolines; The corresponding aminoquinazolines, and certain other new 4-quinazolines. J. Am. Chem. Soc. 1912, 34, 524–534. [Google Scholar]
- Hepworth, W. Quinazoline derivatives. Great Britain Patent 857362, 1960. [Chem. Abstr. 1961, 55, 76248]. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maillard-Boyer, M.; Castera-Ducros, C.; Verhaeghe, P.; Sifredi, F.; Rathelot, P.; Vanelle, P. Access to Original Vinylic Chlorides in the Quinazoline Series via a Monoelectronic Transfer Reaction Approach. Molecules 2010, 15, 2719-2729. https://doi.org/10.3390/molecules15042719
Maillard-Boyer M, Castera-Ducros C, Verhaeghe P, Sifredi F, Rathelot P, Vanelle P. Access to Original Vinylic Chlorides in the Quinazoline Series via a Monoelectronic Transfer Reaction Approach. Molecules. 2010; 15(4):2719-2729. https://doi.org/10.3390/molecules15042719
Chicago/Turabian StyleMaillard-Boyer, Martine, Caroline Castera-Ducros, Pierre Verhaeghe, France Sifredi, Pascal Rathelot, and Patrice Vanelle. 2010. "Access to Original Vinylic Chlorides in the Quinazoline Series via a Monoelectronic Transfer Reaction Approach" Molecules 15, no. 4: 2719-2729. https://doi.org/10.3390/molecules15042719
APA StyleMaillard-Boyer, M., Castera-Ducros, C., Verhaeghe, P., Sifredi, F., Rathelot, P., & Vanelle, P. (2010). Access to Original Vinylic Chlorides in the Quinazoline Series via a Monoelectronic Transfer Reaction Approach. Molecules, 15(4), 2719-2729. https://doi.org/10.3390/molecules15042719