µ-Conotoxins as Leads in the Development of New Analgesics

Abstract
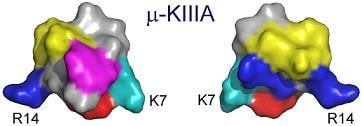
:
{kind=link}
{kind=link}
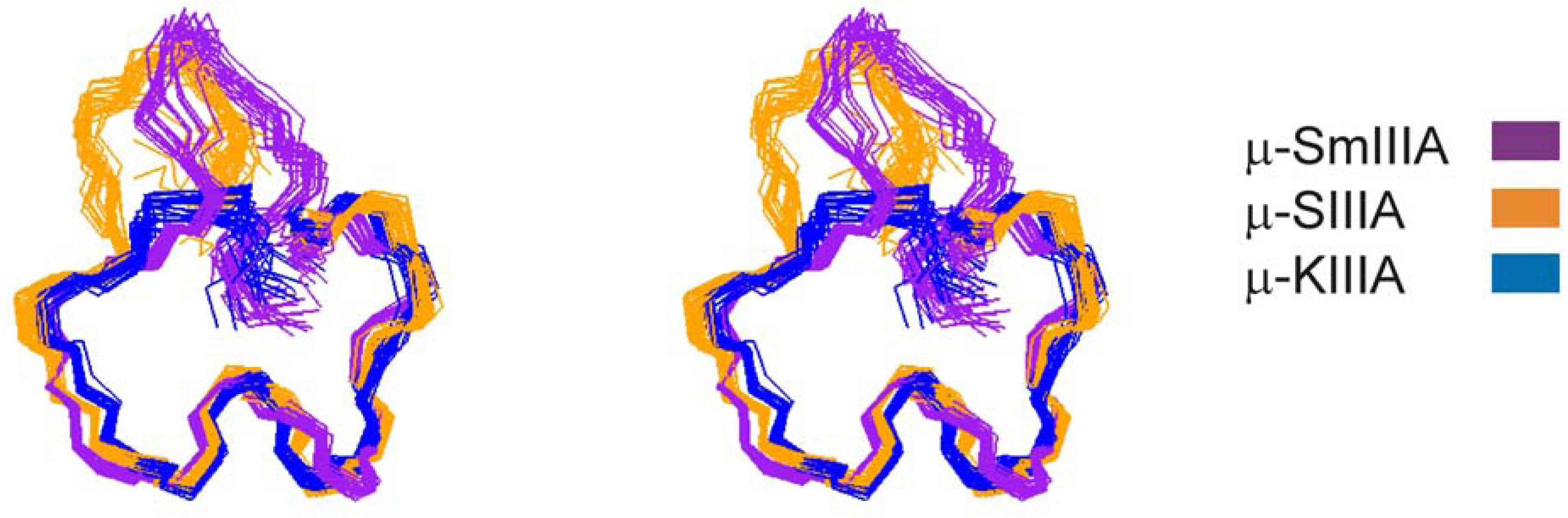{kind=link}
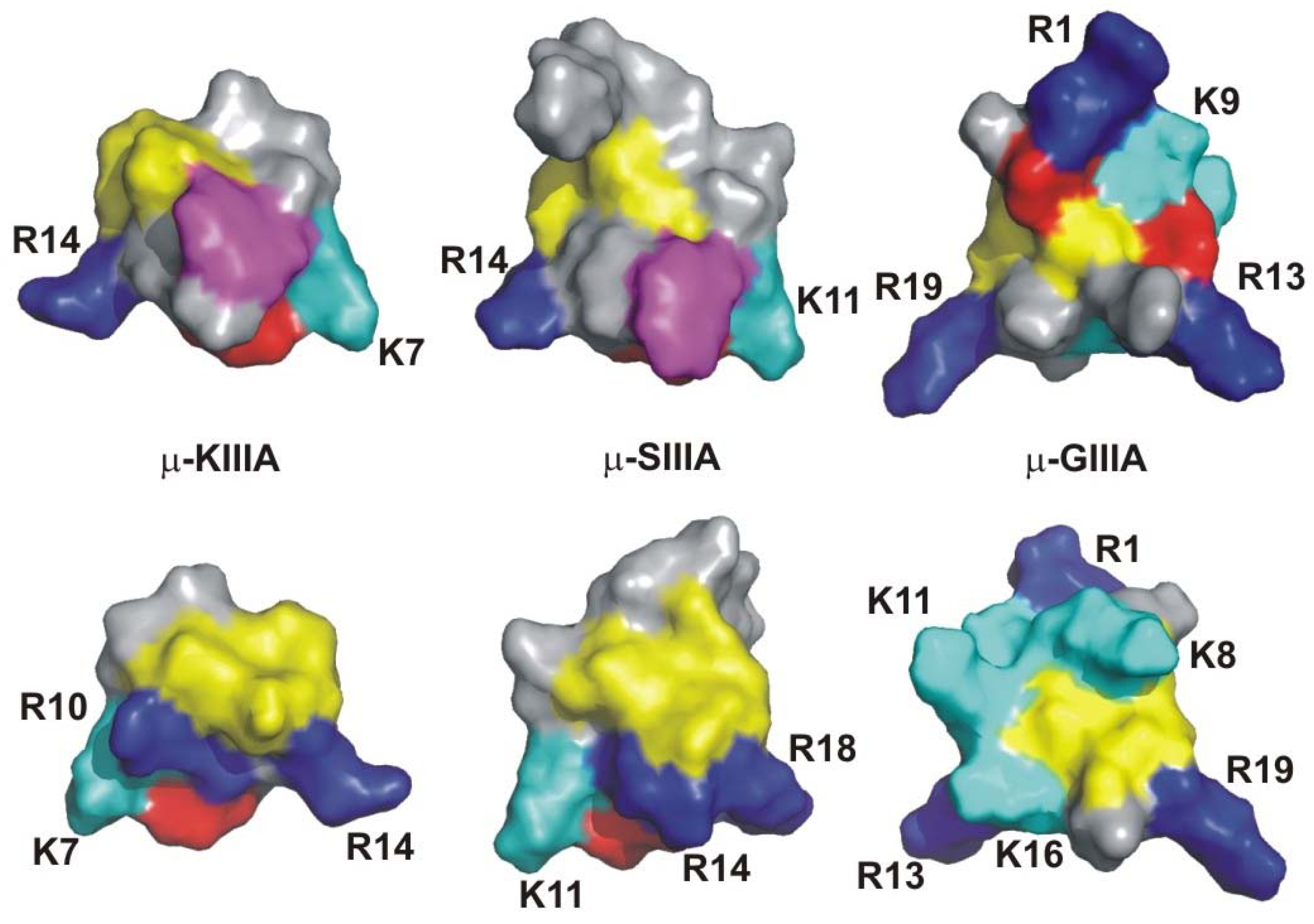{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. μ-Conotoxins

3. Structures and Structure-Function Relationships


3.1. Key Residues of μ-KIIIA Lie on an α-Helix
4. Voltage-Gated Sodium Channels and Pain
5. Exploiting the Therapeutic Potential of μ-Conotoxins
5.1. Peptides as Drugs
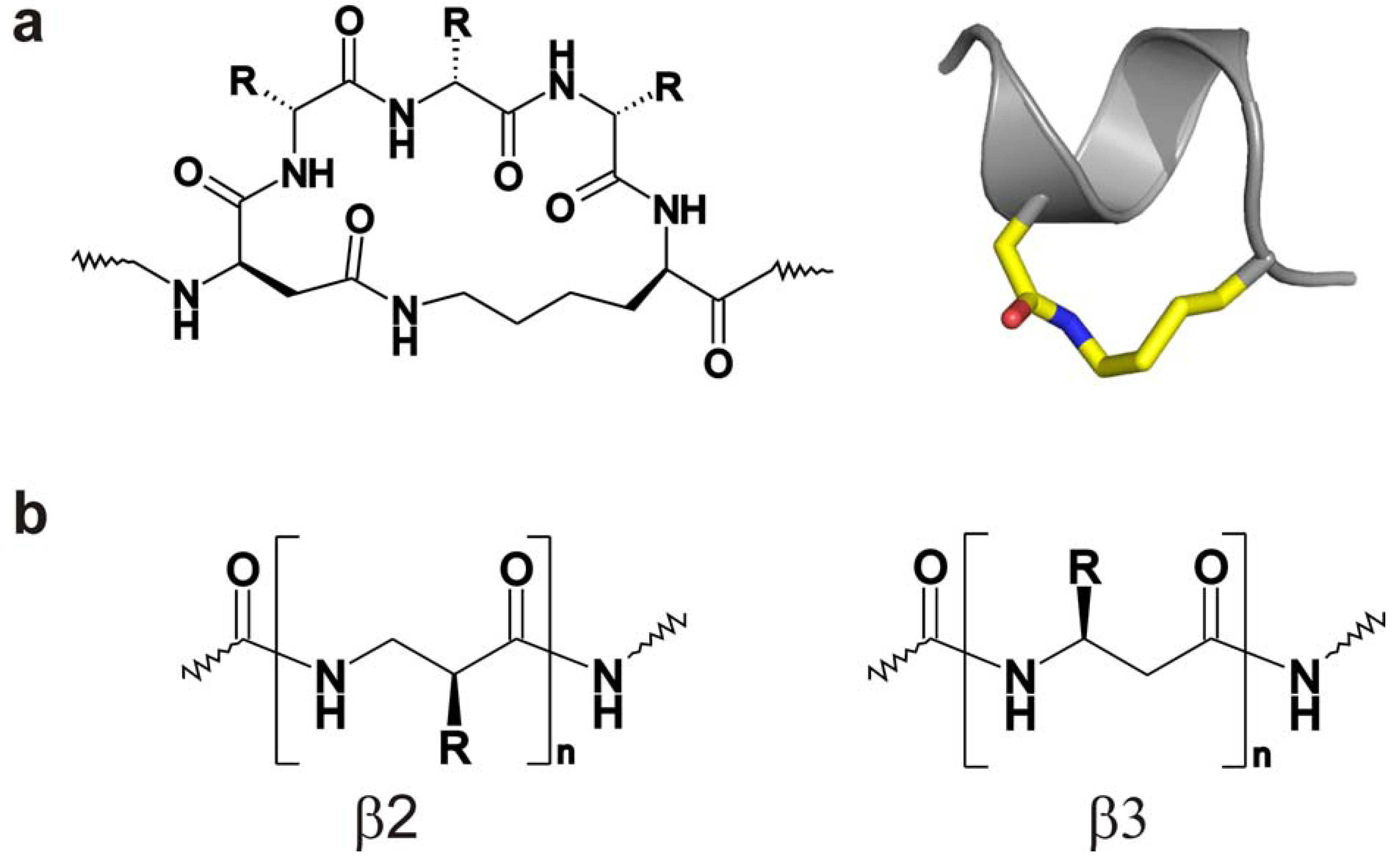
5.2. Truncated and Stabilized Peptides

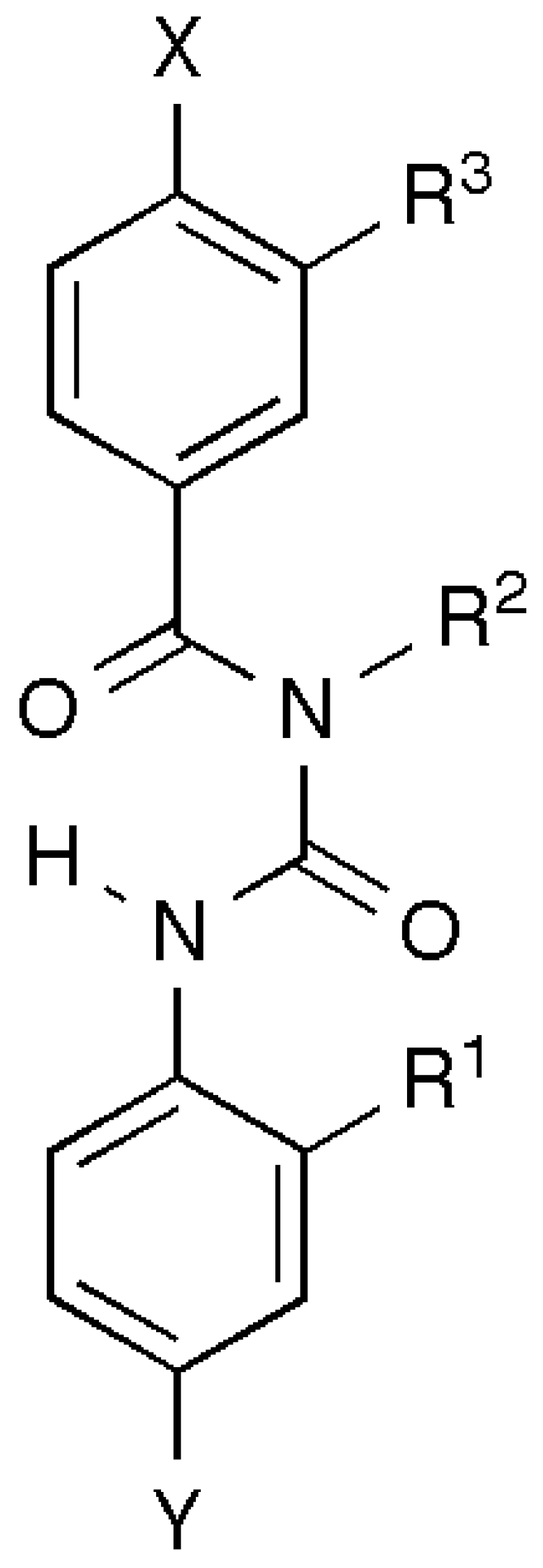
5.3. Peptide Mimetics

6. Conclusions
Acknowledgements
References and Notes
- French, R.J.; Terlau, H. Sodium channel toxins--receptor targeting and therapeutic potential. Curr. Med. Chem. 2004, 11, 3053–3064. [Google Scholar] [CrossRef]
- Cummins, T.R.; Sheets, P.L.; Waxman, S.G. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain 2007, 131, 243–257. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. From genes to pain: NaV1.7 and human pain disorders. Trends Neurosci. 2007, 30, 555–563. [Google Scholar] [CrossRef]
- Catterall, W. A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [Green Version]
- Norton, R.S.; Olivera, B.M. Conotoxins down under. Toxicon 2006, 48, 780–798. [Google Scholar]
- Cestele, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Catterall, W.A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Goldin, A.L. Resurgence of sodium channel research. Annu. Rev. Physiol. 2001, 63, 871–894. [Google Scholar] [CrossRef]
- Moczydlowski, E.; Olivera, B.M.; Gray, W.R.; Strichartz, G.R. Discrimination of muscle and neuronal Na-channel subtypes by binding competition between [3H]saxitoxin and μ-conotoxins. Proc. Natl. Acad. Sci. USA 1986, 83, 5321–5325. [Google Scholar] [CrossRef]
- Chen, L.Q.; Chahine, M.; Kallen, R.G.; Barchi, R.L.; Horn, R. Chimeric study of sodium channels from rat skeletal and cardiac muscle. FEBS Lett. 1992, 309, 253–257. [Google Scholar] [CrossRef]
- Stephan, M.M.; Potts, J.F.; Agnew, W.S. The microI skeletal muscle sodium channel: mutation E403Q eliminates sensitivity to tetrodotoxin but not to μ-conotoxins GIIIA and GIIIB. J. Membr. Biol. 1994, 137, 1–8. [Google Scholar]
- Dudley, S.C., Jr.; Todt, H.; Lipkind, G.; Fozzard, H.A. A μ-conotoxin-insensitive Na+ channel mutant: possible localization of a binding site at the outer vestibule. Biophys. J. 1995, 69, 1657–1665. [Google Scholar] [CrossRef]
- Chahine, M.; Sirois, J.; Marcotte, P.; Chen, L.; Kallen, R.G. Extrapore residues of the S5-S6 loop of domain 2 of the voltage-gated skeletal muscle sodium channel (rSkM1) contribute to the μ-conotoxin GIIIA binding site. Biophys. J. 1998, 75, 236–246. [Google Scholar]
- Terlau, H.; Olivera, B.M. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Corpuz, G.P.; Jacobsen, R.B.; Jimenez, E.C.; Watkins, M.; Walker, C.; Colledge, C.; Garrett, J.E.; McDougal, O.; Li, W.; Gray, W.R.; Hillyard, D.R.; Rivier, J.; McIntosh, J.M.; Cruz, L.J.; Olivera, B.M. Definition of the M-conotoxin superfamily: characterization of novel peptides from molluscivorous Conus venoms. Biochemistry 2005, 44, 8176–8186. [Google Scholar]
- Jacob, R.B.; McDougal, O.M. The M-superfamily of conotoxins: a review. Cell Mol. Life Sci. 2010, 67, 17–27. [Google Scholar] [CrossRef]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M.; Zeikus, R.D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar]
- Lewis, R.J.; Schroeder, C.I.; Ekberg, J.; Nielsen, K.J.; Loughnan, M.; Thomas, L.; Adams, D.A.; Drinkwater, R.; Adams, D.J.; Alewood, P.F. Isolation and structure-activity of μ-conotoxin TIIIA, a potent inhibitor of tetrodotoxin-sensitive voltage-gated sodium channels. Mol. Pharmacol. 2007, 71, 676–685. [Google Scholar]
- Shon, K.J.; Olivera, B.M.; Watkins, M.; Jacobsen, R.B.; Gray, W.R.; Floresca, C.Z.; Cruz, L.J.; Hillyard, D.R.; Brink, A.; Terlau, H.; Yoshikami, D. µ-Conotoxin PIIIA, a new peptide for discriminating among tetrodotoxin-sensitive Na channel subtypes. J. Neurosci. 1998, 18, 4473–4481. [Google Scholar]
- Walewska, A.; Skalicky, J.J.; Davis, D.R.; Zhang, M.M.; Lopez-Vera, E.; Watkins, M.; Han, T.S.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. NMR-based mapping of disulfide bridges in cysteine-rich peptides: application to the μ-conotoxin SxIIIA. J. Am. Chem. Soc. 2008, 130, 14280–14286. [Google Scholar]
- Bulaj, G.; West, P.J.; Garrett, J.E.; Watkins, M.; Zhang, M.M.; Norton, R.S.; Smith, B.J.; Yoshikami, D.; Olivera, B.M. Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry 2005, 44, 7259–7265. [Google Scholar]
- Schroeder, C.I.; Ekberg, J.; Nielsen, K.J.; Adams, D.; Loughnan, M.L.; Thomas, L.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Neuronally micro-conotoxins from Conus striatus utilize an α-helical motif to target mammalian sodium channels. J. Biol. Chem. 2008, 283, 21621–21628. [Google Scholar]
- West, P.J.; Bulaj, G.; Garrett, J.E.; Olivera, B.M.; Yoshikami, D. µ-conotoxin SmIIIA, a potent inhibitor of tetrodotoxin-resistant sodium channels in amphibian sympathetic and sensory neurons. Biochemistry 2002, 41, 15388–15393. [Google Scholar] [CrossRef]
- Holford, M.; Zhang, M.M.; Gowd, K.H.; Azam, L.; Green, B.R.; Watkins, M.; Ownby, J.P.; Yoshikami, D.; Bulaj, G.; Olivera, B.M. Pruning nature: Biodiversity-derived discovery of novel sodium channel blocking conotoxins from Conus bullatus. Toxicon 2009, 53, 90–98. [Google Scholar] [CrossRef]
- Zhang, M.M.; Fiedler, B.; Green, B.R.; Catlin, P.; Watkins, M.; Garrett, J.E.; Smith, B.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Structural and functional diversities among μ-conotoxins targeting TTX-resistant sodium channels. Biochemistry 2006, 45, 3723–3732. [Google Scholar]
- Gray, W.R.; Olivera, B.M.; Cruz, L.J. Peptide toxins from venomous Conus snails. Annu. Rev. Biochem. 1988, 57, 665–700. [Google Scholar]
- Becker, S.; Prusak-Sochaczewski, E.; Zamponi, G.; Beck-Sickinger, A.G.; Gordon, R.D.; French, R.J. Action of derivatives of μ-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry 1992, 31, 8229–8238. [Google Scholar] [CrossRef]
- Cruz, L.J.; Kupryszewski, G.; LeCheminant, G.W.; Gray, W.R.; Olivera, B.M.; Rivier, J. μ-conotoxin GIIIA, a peptide ligand for muscle sodium channels: chemical synthesis, radiolabeling, and receptor characterization. Biochemistry 1989, 28, 3437–3442. [Google Scholar] [CrossRef]
- Sato, K.; Ishida, Y.; Wakamatsu, K.; Kato, R.; Honda, H.; Ohizumi, Y.; Nakamura, H.; Ohya, M.; Lancelin, J.M.; Kohda, D. Active site of μ-conotoxin GIIIA, a peptide blocker of muscle sodium channels. J. Biol. Chem. 1991, 266, 16989–16991. [Google Scholar]
- Chahine, M.; Chen, L.Q.; Fotouhi, N.; Walsky, R.; Fry, D.; Santarelli, V.; Horn, R.; Kallen, R.G. Characterizing the μ-conotoxin binding site on voltage-sensitive sodium channels with toxin analogs and channel mutations. Receptors Channels 1995, 3, 161–174. [Google Scholar]
- French, R.J.; Prusak-Sochaczewski, E.; Zamponi, G.W.; Becker, S.; Kularatna, A.S.; Horn, R. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron 1996, 16, 407–413. [Google Scholar] [CrossRef]
- Hui, C.; Goto, A.; Yamada, K.; Yagi, N.; Nagoshi, H.; Sasabe, M.; Omata, M. Modulation of vascular calcium channel activity in response to acute volume expansion in rats. Life Sci. 1996, 58, 359–366. [Google Scholar]
- Li, R.A.; Sato, K.; Kodama, K.; Kohno, T.; Xue, T.; Tomaselli, G.F.; Marban, E. Charge conversion enables quantification of the proximity between a normally-neutral μ-conotoxin (GIIIA) site and the Na+ channel pore. FEBS Lett. 2002, 511, 159–164. [Google Scholar] [CrossRef]
- Hui, K.; Lipkind, G.; Fozzard, H.A.; French, R.J. Electrostatic and steric contributions to block of the skeletal muscle sodium channel by μ-conotoxin. J. Gen. Physiol. 2002, 119, 45–54. [Google Scholar] [CrossRef]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed; Sinauer Associates,Inc.: Sunderland, MA, USA, 2001. [Google Scholar]
- Safo, P.; Rosenbaum, T.; Shcherbatko, A.; Choi, D.Y.; Han, E.; Toledo-Aral, J.J.; Olivera, B.M.; Brehm, P.; Mandel, G. Distinction among neuronal subtypes of voltage-activated sodium channels by μ-conotoxin PIIIA. J. Neurosci. 2000, 20, 76–80. [Google Scholar]
- Nielsen, K.J.; Watson, M.; Adams, D.J.; Hammarstrom, A.K.; Gage, P.W.; Hill, J.M.; Craik, D.J.; Thomas, L.; Adams, D.; Alewood, P.F.; Lewis, R.J. Solution structure of μ-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. J. Biol. Chem. 2002, 277, 27247–27255. [Google Scholar]
- Wang, C.Z.; Zhang, H.; Jiang, H.; Lu, W.; Zhao, Z.Q.; Chi, C.W. A novel conotoxin from Conus striatus, µ-SIIIA, selectively blocking rat tetrodotoxin-resistant sodium channels. Toxicon 2006, 47, 122–132. [Google Scholar] [CrossRef]
- Zhang, M.M.; Green, B.R.; Catlin, P.; Fiedler, B.; Azam, L.; Chadwick, A.; Terlau, H.; McArthur, J.R.; French, R.J.; Gulyas, J.; Rivier, J.E.; Smith, B.J.; Norton, R.S.; Olivera, B.M.; Yoshikami, D.; Bulaj, G. Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007, 282, 30699–30706. [Google Scholar]
- Khoo, K.K.; Feng, Z.P.; Smith, B.J.; Zhang, M.M.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure of the analgesic μ-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry 2009, 48, 1210–1219. [Google Scholar] [CrossRef]
- Yao, S.; Zhang, M.M.; Yoshikami, D.; Azam, L.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure, dynamics, and selectivity of the sodium channel blocker μ-conotoxin SIIIA. Biochemistry 2008, 47, 10940–10949. [Google Scholar] [CrossRef]
- Keizer, D.W.; West, P.J.; Lee, E.F.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structural basis for tetrodotoxin-resistant sodium channel binding by μ-conotoxin SmIIIA. J. Biol. Chem. 2003, 278, 46805–46813. [Google Scholar]
- Wakamatsu, K.; Kohda, D.; Hatanaka, H.; Lancelin, J.M.; Ishida, Y.; Oya, M.; Nakamura, H.; Inagaki, F.; Sato, K. Structure-activity relationships of μ-conotoxin GIIIA: structure determination of active and inactive sodium channel blocker peptides by NMR and simulated annealing calculations. Biochemistry 1992, 31, 12577–12584. [Google Scholar]
- Lancelin, J.M.; Kohda, D.; Tate, S.; Yanagawa, Y.; Abe, T.; Satake, M.; Inagaki, F. Tertiary structure of conotoxin GIIIA in aqueous solution. Biochemistry 1991, 30, 6908–6916. [Google Scholar]
- Ott, K.H.; Becker, S.; Gordon, R.D.; Ruterjans, H. Solution structure of μ-conotoxin GIIIA analysed by 2D-NMR and distance geometry calculations. FEBS Lett. 1991, 278, 160–166. [Google Scholar] [CrossRef]
- Hill, J.M.; Alewood, P.F.; Craik, D.J. Three-dimensional solution structure of μ-conotoxin GIIIB, a specific blocker of skeletal muscle sodium channels. Biochemistry 1996, 35, 8824–8835. [Google Scholar] [CrossRef]
- Bayrhuber, M.; Vijayan, V.; Ferber, M.; Graf, R.; Korukottu, J.; Imperial, J.; Garrett, J.E.; Olivera, B.M.; Terlau, H.; Zweckstetter, M.; Becker, S. Conkunitzin-S1 is the first member of a new Kunitz-type neurotoxin family. Structural and functional characterization. J. Biol. Chem. 2005, 280, 23766–23770. [Google Scholar]
- Zhang, M.M.; McArthur, J.R.; Azam, L.; Bulaj, G.; Olivera, B.M.; French, R.J.; Yoshikami, D. Synergistic and antagonistic interactions between tetrodotoxin and μ-conotoxin in blocking voltage-gated sodium channels. Channels (Austin) 2009, 3, 32–38. [Google Scholar] [CrossRef]
- Krafte, D.S.; Bannon, A.W. Sodium channels and nociception: recent concepts and therapeutic opportunities. Curr. Opin. Pharmacol. 2008, 8, 50–56. [Google Scholar]
- Priest, B.T. Future potential and status of selective sodium channel blockers for the treatment of pain. Curr. Opin. Drug Discov. Devel. 2009, 12, 682–692. [Google Scholar]
- Amaya, F.; Wang, H.; Costigan, M.; Allchorne, A.J.; Hatcher, J.P.; Egerton, J.; Stean, T.; Morisset, V.; Grose, D.; Gunthorpe, M.J.; Chessell, I.P.; Tate, S.; Green, P.J.; Woolf, C.J. The voltage-gated sodium channel NaV1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 2006, 26, 12852–12860. [Google Scholar]
- Strickland, I.T.; Martindale, J.C.; Woodhams, P.L.; Reeve, A.J.; Chessell, I.P.; McQueen, D.S. Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain 2008, 12, 564–572. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; Al-Gazali, L.; Hamamy, H.; Valente, E.M.; Gorman, S.; Williams, R.; McHale, D.P.; Wood, J.N.; Gribble, F.M.; Woods, C.G. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar]
- Waxman, S.G.; Kocsis, J.D.; Black, J.A. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J. Neurophysiol. 1994, 72, 466–470. [Google Scholar]
- Hains, B.C.; Saab, C.Y.; Klein, J.P.; Craner, M.J.; Waxman, S.G. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J. Neurosci. 2004, 24, 4832–4839. [Google Scholar] [CrossRef]
- Hong, S.; Morrow, T.J.; Paulson, P.E.; Isom, L.L.; Wiley, J.W. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 2004, 279, 29341–29350. [Google Scholar]
- Garry, E.M.; Delaney, A.; Anderson, H.A.; Sirinathsinghji, E.C.; Clapp, R.H.; Martin, W.J.; Kinchington, P.R.; Krah, D.L.; Abbadie, C.; Fleetwood-Walker, S.M. Varicella zoster virus induces neuropathic changes in rat dorsal root ganglia and behavioral reflex sensitisation that is attenuated by gabapentin or sodium channel blocking drugs. Pain 2005, 118, 97–111. [Google Scholar]
- Thakor, D.K.; Lin, A.; Matsuka, Y.; Meyer, E.M.; Ruangsri, S.; Nishimura, I.; Spigelman, I. Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol. Pain 2009, 5, 14. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Honore, P.; Shieh, C.C.; Chapman, M.; Joshi, S.; Zhang, X.F.; Kort, M.; Carroll, W.; Marron, B.; Atkinson, R.; Thomas, J.; Liu, D.; Krambis, M.; Liu, Y.; McGaraughty, S.; Chu, K.; Roeloffs, R.; Zhong, C.; Mikusa, J.P.; Hernandez, G.; Gauvin, D.; Wade, C.; Zhu, C.; Pai, M.; Scanio, M.; Shi, L.; Drizin, I.; Gregg, R.; Matulenko, M.; Hakeem, A.; Gross, M.; Johnson, M.; Marsh, K.; Wagoner, P.K.; Sullivan, J.P.; Faltynek, C.R.; Krafte, D.S. A-803467, a potent and selective NaV1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 2007, 104, 8520–8525. [Google Scholar]
- Green, B.R.; Catlin, P.; Zhang, M.M.; Fiedler, B.; Bayudan, W.; Morrison, A.; Norton, R.S.; Smith, B.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Conotoxins containing nonnatural backbone spacers: cladistic-based design, chemical synthesis, and improved analgesic activity. Chem Biol 2007, 14, 399–407. [Google Scholar] [CrossRef]
- Loffet, A. Peptides as drugs: is there a market? J. Pept. Sci. 2002, 8, 1–7. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Kahan, B.D. Forty years of publication of transplantation proceedings--the second decade: the cyclosporine revolution. Transplant Proc. 2009, 41, 1423–1437. [Google Scholar] [CrossRef]
- Oberg, K. Somatostatin analog octreotide LAR in gastro-entero-pancreatic tumors. Expert Rev. Anticancer Ther. 2009, 9, 557–566. [Google Scholar] [CrossRef]
- Aghi, M.; Blevins, L.S., Jr. Recent advances in the treatment of acromegaly. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 304–307. [Google Scholar] [CrossRef]
- Varkony, H.; Weinstein, V.; Klinger, E.; Sterling, J.; Cooperman, H.; Komlosh, T.; Ladkani, D.; Schwartz, R. The glatiramoid class of immunomodulator drugs. Expert Opin. Pharmacother. 2009, 10, 657–668. [Google Scholar] [CrossRef]
- Chia, C.W.; Egan, J.M. Incretin-based therapies in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab 2008, 93, 3703–3716. [Google Scholar]
- Miljanich, G.P. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef]
- Norton, R.S. Structure and activity of N-methylated peptides. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A.B., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Volume 5, in press. [Google Scholar]
- Pennington, M.W.; Beeton, C.; Galea, C.A.; Smith, B.J.; Chi, V.; Monaghan, K.P.; Garcia, A.; Rangaraju, S.; Giuffrida, A.; Plank, D.; Crossley, G.; Nugent, D.; Khaytin, I.; Lefievre, Y.; Peshenko, I.; Dixon, C.; Chauhan, S.; Orzel, A.; Inoue, T.; Hu, X.; Moore, R.V.; Norton, R.S.; Chandy, K.G. Engineering a stable and selective peptide blocker of the Kv1.3 channel in T lymphocytes. Mol. Pharmacol. 2009, 75, 762–773. [Google Scholar]
- Beeton, C.; Pennington, M.W.; Norton, R.S. Analogs of the sea anemone potassium channel blocker ShK for the treatment of autoimmune diseases. Curr. Immunol. Rev. 2010, in press. [Google Scholar]
- Beeton, C.; Smith, B.J.; Sabo, J.K.; Crossley, G.; Nugent, D.; Khaytin, I.; Chi, V.; Chandy, K.G.; Pennington, M.W.; Norton, R.S. The D-diastereomer of ShK toxin selectively blocks voltage-gated K+ channels and inhibits T lymphocyte proliferation. J. Biol. Chem. 2008, 283, 988–997. [Google Scholar]
- des Rieux, A.; Fievez, V.; Garinot, M.; Schneider, Y.J.; Preat, V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control. Release 2006, 116, 1–27. [Google Scholar]
- Antosova, Z.; Mackova, M.; Kral, V.; Macek, T. Therapeutic application of peptides and proteins: parenteral forever? Trends Biotechnol. 2009, 27, 628–635. [Google Scholar]
- Yang, B.B.; Lum, P.K.; Hayashi, M.M.; Roskos, L.K. Polyethylene glycol modification of filgrastim results in decreased renal clearance of the protein in rats. J. Pharm. Sci. 2004, 93, 1367–1373. [Google Scholar] [CrossRef]
- Bailon, P.; Won, C.Y. PEG-modified biopharmaceuticals. Expert Opin. Drug Deliv. 2009, 6, 1–16. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Daly, N.L.; Nevin, S.T.; Adams, D.J.; Craik, D.J.; Alewood, P.F. α-selenoconotoxins, a new class of potent a7 neuronal nicotinic receptor antagonists. J. Biol. Chem. 2006, 281, 14136–14143. [Google Scholar]
- MacRaild, C.A.; Illesinghe, J.; van Lierop, B.J.; Townsend, A.L.; Chebib, M.; Livett, B.G.; Robinson, A.J.; Norton, R.S. Structure and activity of (2,8)-dicarba-(3,12)-cystino a-ImI, an a-conotoxin containing a nonreducible cystine analogue. J. Med. Chem. 2009, 52, 755–762. [Google Scholar] [CrossRef]
- Muttenthaler, M.; Nevin, S.T.; Grishin, A.A.; Ngo, S.T.; Choy, P.T.; Daly, N.L.; Hu, S.H.; Armishaw, C.J.; Wang, C.I.; Lewis, R.J.; Martin, J.L.; Noakes, P.G.; Craik, D.J.; Adams, D.J.; Alewood, P.F. Solving the a-conotoxin folding problem: efficient seleniuµ-directed on-resin generation of more potent and stable nicotinic acetylcholine receptor antagonists. J. Am. Chem. Soc. 132, 3514–3522.
- Walewska, A.; Zhang, M.M.; Skalicky, J.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Integrated oxidative folding of cysteine/selenocysteine containing peptides: improving chemical synthesis of conotoxins. Angew. Chem. Int. Ed. Engl. 2009, 48, 2221–2224. [Google Scholar] [CrossRef]
- Osapay, G.; Prokai, L.; Kim, H.S.; Medzihradszky, K.F.; Coy, D.H.; Liapakis, G.; Reisine, T.; Melacini, G.; Zhu, Q.; Wang, S.H.; Mattern, R.H.; Goodman, M. Lanthionine-somatostatin analogs: synthesis, characterization, biological activity, and enzymatic stability studies. J. Med. Chem. 1997, 40, 2241–2251. [Google Scholar]
- Goodman, M.; Zapf, C.; Rew, Y. New reagents, reactions, and peptidomimetics for drug design. Biopolymers 2001, 60, 229–245. [Google Scholar] [CrossRef]
- Khoo, K.K.; Norton, R.S. Role of disulfide bonds in peptide and protein conformation. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A.B., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Volume 5, in press. [Google Scholar]
- Gallagher, M.J.; Blumenthal, K.M. Cloning and expression of wild-type and mutant forms of the cardiotonic polypeptide anthopleurin B. J. Biol. Chem. 1992, 267, 13958–13963. [Google Scholar]
- Moran, Y.; Cohen, L.; Kahn, R.; Karbat, I.; Gordon, D.; Gurevitz, M. Expression and mutagenesis of the sea anemone toxin Av2 reveals key amino acid residues important for activity on voltage-gated sodium channels. Biochemistry 2006, 45, 8864–8873. [Google Scholar]
- Moran, Y.; Kahn, R.; Cohen, L.; Gur, M.; Karbat, I.; Gordon, D.; Gurevitz, M. Molecular analysis of the sea anemone toxin Av3 reveals selectivity to insects and demonstrates the heterogeneity of receptor site-3 on voltage-gated Na+ channels. Biochem. J. 2007, 406, 41–48. [Google Scholar]
- Stehling, E.G.; da Silveira, W.D.; Campos, T.A.; Brocchi, M.; Pertinhez, T.A.; Spisni, A. Development of a bacterial cloning vector for expression of scorpion toxins for biotechnological studies. Protein Expr. Purif. 2008, 57, 88–94. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Gardsvoll, H.; Hirschberg, D.; Nielbo, S.; Mayasundari, A.; Peterson, C.B.; Jansson, A.; Jorgensen, T.J.; Poulsen, F.M.; Ploug, M. Solution structure of recombinant somatomedin B domain from vitronectin produced in Pichia pastoris. Protein Sci. 2007, 16, 1934–1945. [Google Scholar] [CrossRef]
- Meta, A.; Nakatake, H.; Imamura, T.; Nozaki, C.; Sugimura, K. High-yield production and characterization of biologically active recombinant aprotinin expressed in Saccharomyces cerevisiae. Protein Expr. Purif. 2009, 66, 22–27. [Google Scholar] [CrossRef]
- Brondyk, W.H. Selecting an appropriate method for expressing a recombinant protein. Meth. Enzymol. 2009, 463, 131–147. [Google Scholar] [CrossRef]
- Han, T.S.; Zhang, M.M.; Walewska, A.; Gruszczynski, P.; Robertson, C.R.; Cheatham, T.E., 3rd; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Structurally minimized μ-conotoxin analogues as sodium channel blockers: implications for designing conopeptide-based therapeutics. ChemMedChem 2009, 4, 406–414. [Google Scholar] [CrossRef]
- Andrews, M.J.I.; Tabor, A.B. Forming stable helical peptides using natural and artificial amino acids. Tetrahedron 1999, 55, 11711–11743. [Google Scholar] [CrossRef]
- Felix, A.M.; Heimer, E.P.; Wang, C.T.; Lambros, T.J.; Fournier, A.; Mowles, T.F.; Maines, S.; Campbell, R.M.; Wegrzynski, B.B.; Toome, V.; Fry, D.; Madison, V.S. Synthesis, biological activity and conformational analysis of cyclic GRF analogs. Int. J. Pept. Protein Res. 1988, 32, 441–454. [Google Scholar]
- Houston, M.E., Jr.; Gannon, C.L.; Kay, C.M.; Hodges, R.S. Lactam bridge stabilization of α-helical peptides: ring size, orientation and positional effects. J. Pept. Sci. 1995, 1, 274–282. [Google Scholar] [CrossRef]
- Taylor, J.W. The synthesis and study of side-chain lactaµ-bridged peptides. Biopolymers 2002, 66, 49–75. [Google Scholar] [CrossRef]
- Kirby, D.A.; Britton, K.T.; Aubert, M.L.; Rivier, J.E. Identification of high-potency neuropeptide Y analogues through systematic lactamization. J. Med. Chem. 1997, 40, 210–215. [Google Scholar]
- Yao, S.; Smith-White, M.A.; Potter, E.K.; Norton, R.S. Stabilization of the helical structure of Y2-selective analogues of neuropeptide Y by lactam bridges. J. Med. Chem. 2002, 45, 2310–2318. [Google Scholar]
- Osapay, G.; Taylor, J.W. Multicyclic polypeptide model compounds. 2. Synthesis and conformational properties of a highly α-helical uncosapeptide constrained by three side-chain to side-chain lactam bridges. J. Am. Chem. Soc. 1992, 114, 6966–6973. [Google Scholar] [CrossRef]
- Lanigan, M.D.; Pennington, M.W.; Lefievre, Y.; Rauer, H.; Norton, R.S. Designed peptide analogues of the potassium channel blocker ShK toxin. Biochemistry 2001, 40, 15528–15537. [Google Scholar]
- Yang, B.; Liu, D.; Huang, Z. Synthesis and helical structure of lactam bridged BH3 peptides derived from pro-apoptotic Bcl-2 family proteins. Bioorg. Med. Chem. Lett. 2004, 14, 1403–1406. [Google Scholar] [CrossRef]
- Shepherd, N.E.; Abbenante, G.; Fairlie, D.P. Consecutive cyclic pentapeptide modules form short α-helices that are very stable to water and denaturants. Angew. Chem. Int. Ed. Engl. 2004, 43, 2687–2690. [Google Scholar] [CrossRef]
- Shepherd, N.E.; Hoang, H.N.; Abbenante, G.; Fairlie, D.P. Left- and right-handed α-helical turns in homo- and hetero-chiral helical scaffolds. J. Am. Chem. Soc. 2009, 131, 15877–15886. [Google Scholar]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar]
- Bird, G.H.; Bernal, F.; Pitter, K.; Walensky, L.D. Synthesis and biophysical characterization of stabilized α-helices of BCL-2 domains. Meth. Enzymol. 2008, 446, 369–386. [Google Scholar] [CrossRef]
- Phelan, J.C.; Skelton, N.J.; Braisted, A.C.; McDowell, R.S. A general method for constraining short peptides to an α-helical conformation. J. Am. Chem. Soc. 1997, 119, 455–460. [Google Scholar] [CrossRef]
- Yu, C.; Taylor, J.W. Synthesis and study of peptides with semirigid i and i + 7 side-chain bridges designed for α-helix stabilization. Bioorg. Med. Chem. 1999, 7, 161–175. [Google Scholar] [CrossRef]
- Moellering, R.E.; Cornejo, M.; Davis, T.N.; Del Bianco, C.; Aster, J.C.; Blacklow, S.C.; Kung, A.L.; Gilliland, D.G.; Verdine, G.L.; Bradner, J.E. Direct inhibition of the NOTCH transcription factor complex. Nature 2009, 462, 182–188. [Google Scholar]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: from structure to function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef]
- Seebach, D.; Hook, D.F.; Glattli, A. Helices and other secondary structures of β- and γ-peptides. Biopolymers 2006, 84, 23–37. [Google Scholar] [CrossRef]
- Horne, W.S.; Gellman, S.H. Foldamers with heterogeneous backbones. Acc. Chem. Res. 2008, 41, 1399–1408. [Google Scholar] [CrossRef]
- Sadowsky, J.D.; Fairlie, W.D.; Hadley, E.B.; Lee, H.S.; Umezawa, N.; Nikolovska-Coleska, Z.; Wang, S.; Huang, D.C.; Tomita, Y.; Gellman, S.H. (a/β+a)-peptide antagonists of BH3 domain/Bcl-x(L) recognition: toward general strategies for foldamer-based inhibition of protein-protein interactions. J. Am. Chem. Soc. 2007, 129, 139–154. [Google Scholar]
- Horne, W.S.; Boersma, M.D.; Windsor, M.A.; Gellman, S.H. Sequence-based design of α/β-peptide foldamers that mimic BH3 domains. Angew. Chem. Int. Ed. Engl. 2008, 47, 2853–2856. [Google Scholar]
- Baell, J.B.; Forsyth, S.A.; Gable, R.W.; Norton, R.S.; Mulder, R.J. Design and synthesis of type-III mimetics of ω-conotoxin GVIA. J. Comput. Aided Mol. Des. 2001, 15, 1119–1136. [Google Scholar] [CrossRef]
- Baell, J.B.; Harvey, A.J.; Norton, R.S. Design and synthesis of type-III mimetics of ShK toxin. J. Comput. Aided Mol. Des. 2002, 16, 245–262. [Google Scholar] [CrossRef]
- Harvey, A.J.; Gable, R.W.; Baell, J.B. A three-residue, continuous binding epitope peptidomimetic of ShK toxin as a Kv1.3 inhibitor. Bioorg. Med. Chem. Lett. 2005, 15, 3193–3196. [Google Scholar]
- Baell, J.B.; Duggan, P.J.; Forsyth, S.A.; Lewis, R.J.; Lok, Y.P.; Schroeder, C.I. Synthesis and biological evaluation of nonpeptide mimetics of ω-conotoxin GVIA. Bioorg. Med. Chem. 2004, 12, 4025–4037. [Google Scholar]
- Norton, R.S.; Baell, J.B.; Angus, J.A. Calcium channel blocking polypeptides: structure, function and molecular mimicry. In Calcium Channel Pharmacology; McDonough, S.I., Ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004; pp. 143–181. [Google Scholar]
- Orner, B.P.; Ernst, J.T.; Hamilton, A.D. Toward proteomimetics: terphenyl derivatives as structural and functional mimics of extended regions of an α-helix. J. Am. Chem. Soc. 2001, 123, 5382–5383. [Google Scholar] [CrossRef]
- Kutzki, O.; Park, H.S.; Ernst, J.T.; Orner, B.P.; Yin, H.; Hamilton, A.D. Development of a potent Bcl-x(L) antagonist based on α-helix mimicry. J. Am. Chem. Soc. 2002, 124, 11838–11839. [Google Scholar] [CrossRef]
- Ernst, J.T.; Kutzki, O.; Debnath, A.K.; Jiang, S.; Lu, H.; Hamilton, A.D. Design of a protein surface antagonist based on α-helix mimicry: inhibition of gp41 assembly and viral fusion. Angew. Chem. Int. Ed. Engl. 2002, 41, 278–281. [Google Scholar]
- Cummings, C.G.; Ross, N.T.; Katt, W.P.; Hamilton, A.D. Synthesis and biological evaluation of a 5-6-5 imidazole-phenyl-thiazole based α-helix mimetic. Org. Lett. 2009, 11, 25–28. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Ross, N.T.; Katt, W.P.; Dhar, D.; Lee, G.I.; Hamilton, A.D. Structure and function of benzoylurea-derived α-helix mimetics targeting the Bcl-x(L)/Bak binding interface. ChemMedChem 2009, 4, 649–656. [Google Scholar] [CrossRef]
- Lessene, G.; Smith, B.J.; Gable, R.W.; Baell, J.B. Characterization of the two fundamental conformations of benzoylureas and elucidation of the factors that facilitate their conformational interchange. J. Org. Chem. 2009, 74, 6511–6525. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Norton, R.S. µ-Conotoxins as Leads in the Development of New Analgesics. Molecules 2010, 15, 2825-2844. https://doi.org/10.3390/molecules15042825
Norton RS. µ-Conotoxins as Leads in the Development of New Analgesics. Molecules. 2010; 15(4):2825-2844. https://doi.org/10.3390/molecules15042825
Chicago/Turabian StyleNorton, Raymond S. 2010. "µ-Conotoxins as Leads in the Development of New Analgesics" Molecules 15, no. 4: 2825-2844. https://doi.org/10.3390/molecules15042825
APA StyleNorton, R. S. (2010). µ-Conotoxins as Leads in the Development of New Analgesics. Molecules, 15(4), 2825-2844. https://doi.org/10.3390/molecules15042825