3.2. Synthesis
5-Chloro-2H-chromene-3-carboxaldehyde (
4). A mixture of 6-chlorosalicylaldehyde
3 [
7] (16.5 g, 0.105 mol), DABCO (5.90 g, 0.053 mol), acrolein (10.5 mL, 0.158 mol), and dioxane (36 mL) was placed in a sealed vial and heated with stirring at 95 °C for 140 minutes. The reaction mixture was cooled to room temperature, diluted with CH
2Cl
2, washed with 10% aqueous HCl and then brine, dried with anhydrous Na
2SO
4, and evaporated. Chromatography on silica using a gradient of 25–50% CH
2Cl
2 in hexanes gave 14.8 g (72%) of
4 as light yellow crystals. An analytical sample was recrystallized from EtOAc/hexanes: m.p. 65.5–66 °C.
1H-NMR (DMSO-
d6) δ 9.69 (s, C
HO, 1H), 7.77 (s, C
H=CCHO, 1H), 7.34 (dd, Ar
H,
J = 8.0, 8.0 Hz, 1H), 7.13 (d, Ar
H,
J = 8.0 Hz, 1H), 6.88 (d, Ar
H,
J = 8.0 Hz, 1H), 4.94 (s, OC
H2, 2H).
13C-NMR (CDCl
3) δ 189.7, 157.1, 137.0, 133.8, 133.0, 132.3, 122.6, 119.2, 115.3, 63.0. HRMS (M+H)
+: calc. for C
10H
8O
2Cl, 195.0207; found, 195.0204.
5-Chloro-2H-chromene-3-carboxylic acid (5). To absolute ethanol (195 mL) in a round-bottomed flask was added a solution of sodium hydroxide (12.2 g, 305 mmol) in water (97 mL). A solution of silver nitrate (27.2 g, 160 mmol) in water (97 mL) was then added dropwise with vigorous stirring. To the resulting suspension of Ag2O was added aldehyde 4 (14.8 g), and the mixture was heated and stirred at 85 °C for 75 minutes. The mixture was cooled to room temperature and the clear supernatant was decanted. The solid was washed with a 1:1 ethanol/water solution (3 × 20 mL), and the washings were combined with the decanted supernatant. Dilution with an excess of 1M aq. HCl gave a voluminous white precipitate which dissolved upon extraction with CH2Cl2. The resulting CH2Cl2 solution was dried with Na2SO4, filtered, and evaporated to give 15.5 g (97%) of 5 as cream-colored fluffy crystals. An analytical sample was recrystallized from EtOAc/hexanes: m.p. 192.5–193 °C. 1H- NMR (DMSO-d6) δ 13.10 (bs, CO2H, 1H), 7.54 (m, CH=CCHO, 1H), 7.28 (dd, ArH, J = 8.2, 8.2 Hz, 1H), 7.09 (dd, ArH, J = 8.2, 1.1 Hz, 1H), 6.87 (ddd, ArH, J = 8.2, 1.1, 1.1 Hz, 1H), 4.92 (s, OCH2, 2H). 13C-NMR (CDCl3) δ 168.7, 156.6, 133.8, 132.3, 132.1, 122.7, 122.5, 119.4, 115.1, 64.0. HRMS (M–H)–: calc. for C10H6O3Cl, 209.0011; found, 209.0012.
5-Chlorochroman-3-carboxylic acid (
6). To a solution of
5 (7.50 g) in 10% aqueous NaOH (193 mL) was added 3% sodium amalgam (103 g) [
15]. The mixture was stirred overnight at room temperature. The supernatant was decanted from the liquid mercury, and the mercury was washed twice with small portions of 10% aq. NaOH. The washings were combined with the supernatant, acidified to a pH of 2 with conc. HCl, and extracted with CH
2Cl
2. The CH
2Cl
2 solution was dried with Na
2SO
4, filtered, and evaporated to give 7.51 g (99%) of
6 as a white crystalline solid. An analytical sample was recrystallized from EtOAc/hexanes: m.p. 129.5–130 °C.
1H-NMR (DMSO-
d6) δ 12.70 (bs, CO
2H, 1H), 7.10 (dd, Ar
H,
J = 8.0, 8.0 Hz, 1H), 6.99 (dd, Ar
H,
J = 8.0, 1.2 Hz, 1H), 6.75 (dd, Ar
H,
J = 8.0, 1.2 Hz, 1H), 4.28 (dd, OC
H2CH,
J = 10.8, 4.3 Hz, 1H), 4.16 (dd, OC
H2CH,
J = 10.8, 7.0 Hz, 1H), 3.05 (m, C
HCO
2H, 1H), 2.90 (d, ArC
H2CH,
J = 6.6 Hz, 2H).
13C-NMR (CDCl
3) δ 178.3, 155.2, 134.6, 127.8, 121.7, 119.0, 115.4, 65.8, 38.2, 25.4. HRMS (M–H)
–: calc. for C
10H
8O
3Cl, 211.0167; found, 211.0166.
5-Chloro-N,N-dimethylchroman-3-carboxamide (7). A mixture of 6 (1.50 g, 7.06 mmol), dimethylamine hydrochloride (1.44 g, 17.6 mmol), HOBt (1.43 g, 10.6 mmol), N-methylmorpholine (3.88 mL, 35.3 mmol), and EDC hydrochloride (2.03 g, 10.6 mmol) in dichloromethane (65 mL) was stirred at room temperature for 50 hours. The reaction mixture was diluted with additional CH2Cl2, and an equal volume of saturated aq. NaHCO3 was added. The CH2Cl2 phase was separated, and the aqueous phase was washed 3× with CH2Cl2. The CH2Cl2 phases were combined, dried with Na2SO4, and evaporated. Chromatography on silica (25–75% EtOAc in hexanes) gave 1.56 g (92%) of 7 as a pale yellow oil. 1H-NMR (DMSO-d6) δ 7.12 (dd, Ar-H, J = 8.2, 8.0 Hz, 1H), 7.01 (dd, Ar-H, J = 8.0, 1.1 Hz, 1H), 6.79 (dd, Ar-H, J = 8.2, 1.1 Hz, 1H), 4.31 (m, OCH2CH, 1H), 3.84 (dd, OCH2CH, J = 10.8, 10.8 Hz, 1H), 3.37-3.30 (m, partly hidden, CHCONMe2, 1H), 3.11 (s, NCH3, 3H), 2.89 (dd, partly hidden, ArCH2CH, J = 16.8, 5.7 Hz, 1H), 2.86 (s, NCH3, 3H), 2.79 (dd, ArCH2CH, J = 16.8, 10.2 Hz, 1H). 13C-NMR (CDCl3) δ 171.7, 155.3, 134.6, 127.6, 121.3, 119.9, 115.2, 67.0, 37.2, 35.6, 35.4, 26.9. HRMS (M+H)+: calc. for C12H15NO2Cl, 240.0786; found, 240.0792.
5-Chloro-N,N-dimethyl-6-nitrochroman-3-carboxamide (8). Sodium nitrate (510 mg, 6.00 mmol) was added to a flask containing trifluoroacetic acid (37.5 mL) at 0 °C. After stirring for 10 minutes, the mixture was cooled to –15 °C (by adding small pieces of dry ice to acetone in a Dewar bath) whereupon the solvent began to freeze. With stirring, a solution of 7 (1.25 g, 5.21 mmol) in trifluoroacetic acid (12.5 mL) was added dropwise. An insulating cover was then placed over the Dewar and the mixture was stirred and allowed to warm very slowly to 18 °C over 12 hours. The resulting red solution was quenched with ice, diluted with water, and extracted with dichloromethane. The dichloromethane phase was washed with water, then 5% aq. KH2PO4 until the pH of the aqueous wash was 4 to 5. The dichloromethane phase was dried with Na2SO4 and evaporated. The resulting residue was purified on silica (25–75% EtOAc in hexanes) to give 510 mg (34%) of 8 as a yellow oil. 1H-NMR (DMSO-d6) δ 7.86 (d, Ar-H, J = 9.0 Hz, 1H), 7.00 (d, Ar-H, J = 9.0 Hz, 1H), 4.39 (m, OCH2CH, 1H), 4.01 (dd, OCH2CH, J = 11.0, 8.8 Hz, 1H), 3.40 (m, CHCO2H, 1H), 3.10 (s, NCH3, 3H), 2.88 (dd, ArCH2CH, J = 17.0, 5.3 Hz, 1H), 2.85 (s, NCH3, 3H), 2.84 (dd, partly hidden, ArCH2CH, J = 17.0, 9.2 Hz, 1H). 13C-NMR (CDCl3) δ 170.8, 158.2, 141.6, 128.7, 124.9, 122.1, 115.4, 67.4, 37.3, 35.7, 34.7, 27.3. The undesired minor ortho isomer, which eluted just before compound 8, was also isolated: 250 mg (17%) of 5-chloro-N,N-dimethyl-8-nitrochroman-3-carboxamide as a pale yellow oil, 1H-NMR (DMSO-d6) δ 7.79 (d, Ar-H, J = 8.8 Hz, 1H), 7.21 (d, Ar-H, J = 8.8 Hz, 1H), 4.48 (m, OCH2CH, 1H), 4.06 (dd, OCH2CH, J = 10.9, 9.1 Hz, 1H), 3.46 (m, CHCO2H, 1H), 3.11 (s, NCH3, 3H), 2.95 (dd, ArCH2CH, J = 17.0, 5.8 Hz, 1H), 2.86 (s, NCH3, 3H), 2.86 (dd, partly hidden, ArCH2CH, J = 16.7, 10.0 Hz, 1H). 13C-NMR (CDCl3) δ 170.5, 149.3, 139.8, 137.6, 124.0, 123.1, 120.6, 67.8, 37.2, 35.7, 34.4, 27.2.
tert-Butyl N-[3-[[3-(dimethylcarbamoyl)-6-nitrochroman-5-yl]amino]propyl]-N-methylcarbamate (9). A mixture of 8 (300 mg, 1.05 mmol), tert-butyl N-(3-aminopropyl)-N-methylcarbamate (989 mg, 5.25 mmol), and N-methylpyrrolidinone (989 mg) was heated in a sealed tube at 75 °C for 22 hours. The reaction mixture was diluted with Et2O (75 mL) and washed with H2O (4 × 75 mL) and 5% aq. KH2PO4 (75 mL), then dried with Na2SO4, filtered, and evaporated. The crude product was purified on silica using a gradient of 40–50% EtOAc in CH2Cl2 to give 399 mg (87%) of 9 as a bright yellow-orange oil. 1H-NMR (DMSO-d6) δ 7.82 (d, Ar-H, J = 9.4 Hz, 1H), 7.20 (b, NH, 1H), 6.35 (d, Ar-H, J = 9.4 Hz, 1H), 4.37 (m, OCH2CH, 1H), 3.93 (dd, OCH2CH,J = 10.6, 10.6 Hz, 1H), 3.25 (m, partly hidden, CHCO2H, 1H), 3.25–3.10 (m, 2 × NCH2, 4H), 3.06 (s, NCH3, 3H), 2.85 (s, NCH3, 3H), 2.80–2.70 (m, partly hidden, ArCH2CH, 2H), 2.71 (s, NCH3, 3H), 1.70 (qu, CH2CH2CH2, J = 6.8 Hz, 2H), 1.34 (s, t-Bu, 9H). 13C-NMR (CDCl3) δ 171.4, 160.1, 155.7, 148.8, 131.6, 126.5, 111.0, 109.3, 79.6, 77.2, 67.4, 45.8, 37.3, 35.7, 35.3, 34.4, 29.4, 28.5, 27.9. HRMS (M+H)+: calc. for C21H33N4O6, 437.2395; found, 437.2401.
tert-Butyl N-[3-[2-amino-8-(dimethylcarbamoyl)-8,9-dihydro-7H-pyrano[2,3-g]benzimidazol-1-yl]- propyl]-N-methylcarbamate (10). To a flask containing 10% palladium on carbon (180 mg) was added a solution of 9 (187 mg, 0.43 mmol) in absolute ethanol (20 mL). The mixture was stirred for 90 minutes under 1 atmosphere of hydrogen at room temperature, at which point 1H-NMR analysis showed complete reduction to the corresponding aniline derivative. The mixture was filtered through Celite. The Celite was washed with three 5-mL aliquots of acetonitrile and all filtrates were combined. To this solution was added cyanogen bromide (49 mg) in acetonitrile (2 mL), and the mixture was stirred at room temperature for 15 hours under argon. The solvent was evaporated, and the residue was dissolved in DMSO (1 mL) and purified by preparative HPLC using a gradient of 5–50% methanol/water/0.1% formic acid over 15 minutes, followed by a ramp to 100% methanol/0.1% formic acid over 3 minutes. The fractions containing the major peak were combined and neutralized with aq. NaHCO3. Most of the methanol was evaporated in vacuo, and the resulting aqueous phase was extracted with CH2Cl2. Drying with Na2SO4, filtration, and evaporation gave 119 mg (64%) of 10 as a nearly colorless oil which solidified into a light tan crystalline solid. An analytical sample was recrystallized from EtOAc/hexanes: m.p. 165.5–166 °C. 1H-NMR (DMSO-d6) δ 6.86 (d, Ar-H, J = 8.4 Hz, 1H), 6.43 (d, Ar-H, J = 8.4 Hz, 1H), 6.09 (bs, Ar-NH2, 2H), 4.23 (dd, OCH2CH, J = 10.6, 2.93 Hz, 1H), 4.03 (bm, NCH2CH2, 2H), 3.78 (dd, OCH2CH, J = 10.4, 10.4 Hz, 1H), 3.35–3.10 (m, major overlap, CHCONMe2, ArCH2CH, CH2NMe, 5H), 3.12 (s, NCH3, 3H), 2.88 (s, NCH3, 3H), 2.76 (s, NCH3, 3H), 1.81 (m, CH2CH2CH2, 2H), 1.33 (bs, t-Bu, 9H). 13C-NMR (DMSO-d6) δ 171.9, 154.9, 148.1, 137.1, 132.0, 114.1, 109.9, 104.5, 78.9, 66.6, 46.2, 45.7, 41.6, 37.2, 35.5, 35.2, 34.1, 29.3, 28.4, 24.3. HRMS (M+H)+: calc. for C22H34N5O4, 432.2605; found, 432.2621.
6-Amino-N,N-dimethylchroman-3-carboxamide (11). Compound 8 (42 mg) was dissolved in absolute ethanol (10 mL) and stirred with 10% palladium on carbon (65 mg) under 1 atm of hydrogen at 50 °C for 3 hours. The mixture was filtered through Celite, and the absorbent was washed several times with ethanol. The combined filtrates were evaporated to give a quantitative yield of 11 as a tan solid. 1H- NMR (DMSO-d6) δ 8.71 (bs, Ar-NH2, 2H), 6.86 (s, Ar-H, 1H), 6.84 (d, Ar-H, J = 8.5 Hz, 1H), 6.73 (d, Ar-H, J = 8.5 Hz, 1H), 4.26 (m, OCH2CH, 1H), 3.81 (dd, OCH2CH, J = 10.9, 10.0 Hz, 1H), 3.23 (m, CHCO2H, 1H), 3.08 (s, NCH3, 3H), 2.86 (dd, partly hidden, ArCH2CH, J = 16.1, 10.6 Hz, 1H), 2.84 (s, NCH3, 3H), 2.79 (dd, ArCH2CH, J = 16.4, 5.4 Hz, 1H).
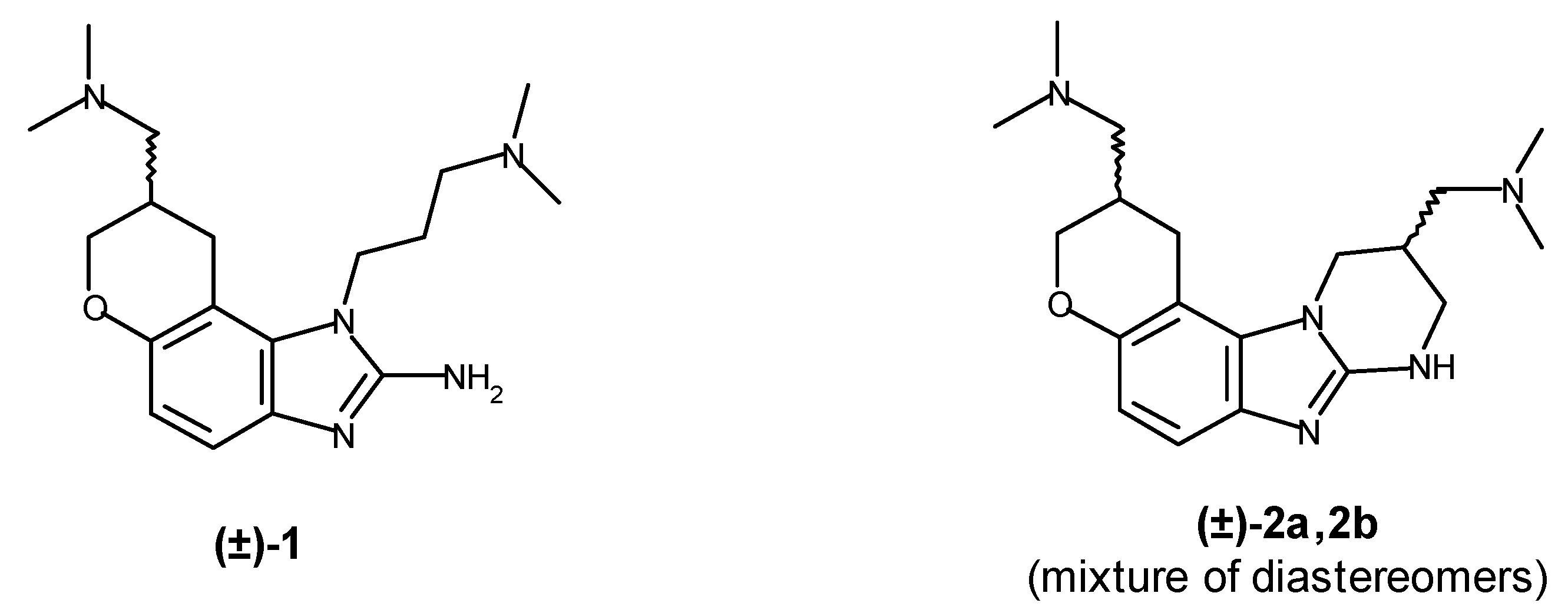
8-(Dimethylaminomethyl)-1-(3-dimethylaminopropyl)-8,9-dihydro-7H-pyrano[3,2-e]benzimidazol-2-amine (1). Compound 10 (100 mg, 0.232 mmol) was dissolved in THF (5 mL) under argon. Lithium aluminum hydride (333 mg, 8.77 mmol) was added and the mixture was stirred at 60 °C for 1.5 hours, then quenched by careful dropwise addition of 3M aq. NaOH. The solids were filtered off and washed with THF. The filtrate and washings were combined, dried with Na2SO4, and evaporated to give a tan solid, which was triturated with a minimal amount of EtOAc to give 68 mg (89%) of 1 as an off-white crystalline solid: m.p. 179.5–180.5 °C. 1H-NMR (DMSO-d6) δ 6.83 (d, Ar-H, J = 8.4 Hz, 1H), 6.39 (d, Ar-H, J = 8.4 Hz, 1H), 6.14 (bs, Ar-NH2, 2H), 4.13 (d, OCH2CH, J = 9.4 Hz, 1H), 4.04 (dd, NCH2CH2, J = 6.9, 6.9 Hz, 2H), 3.72 (dd, OCH2CH, J = 10.2, 7.2 Hz, 1H), 3.16 (dd, ArCH2CH, J = 16.0, 4.5 Hz, 1H), 2.76 (dd, ArCH2CH, J = 16.0, 7.2 Hz, 1H), 2.30-2.20 (m, major overlap, CHCONMe2, 2 × CH2NMe2, 5H), 2.17 (s, NMe2, 6H), 2.16 (s, NMe2, 6H), 1.80 (m, CH2CH2CH2, 2H). 13C-NMR (CDCl3) δ 155.2, 149.1, 136.7, 132.2, 115.1, 110.9, 104.0, 68.5, 61.8, 53.8, 46.0, 44.4, 39.9, 30.6, 29.0, 25.6. HRMS (M+H)+: calc. for C18H30N5O, 332.2445; found, 332.2453.

{kind=link}
{kind=link}
{kind=link}
{kind=link}



