Novel Synthesis and Antitumor Evaluation of Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-Cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide

Abstract
: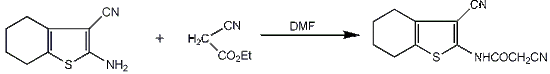
1. Introduction
2. Results and Discussion
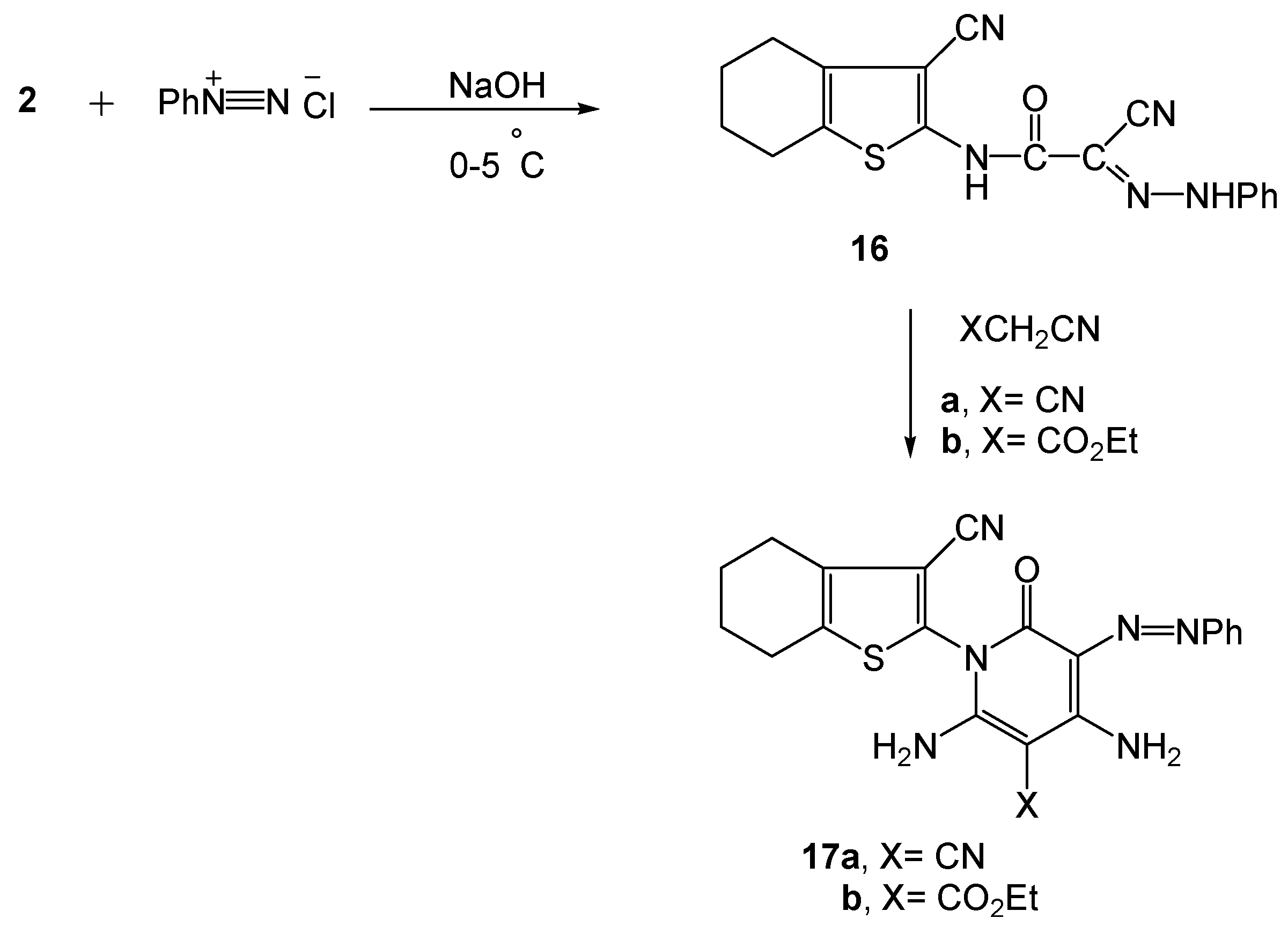
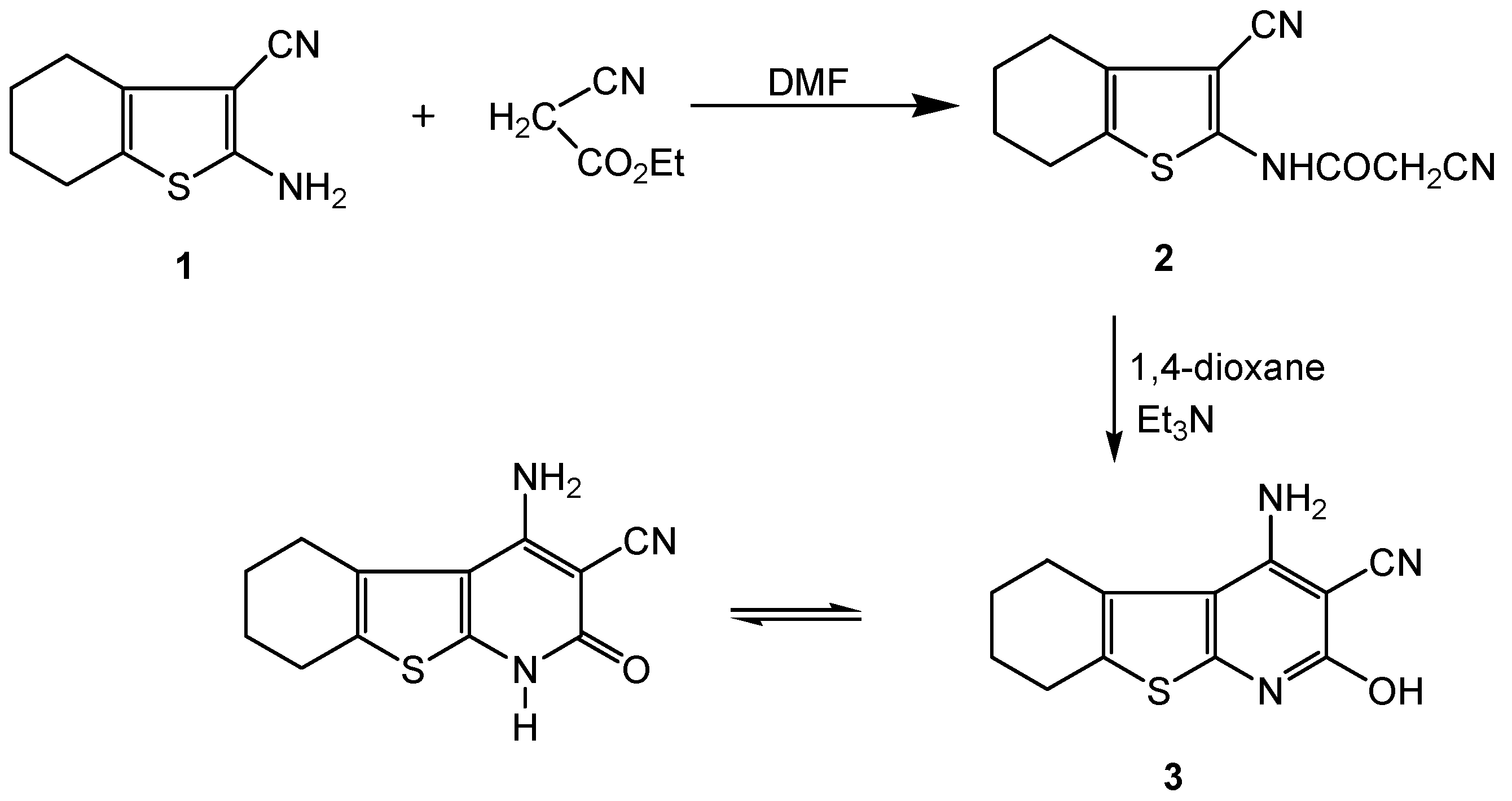
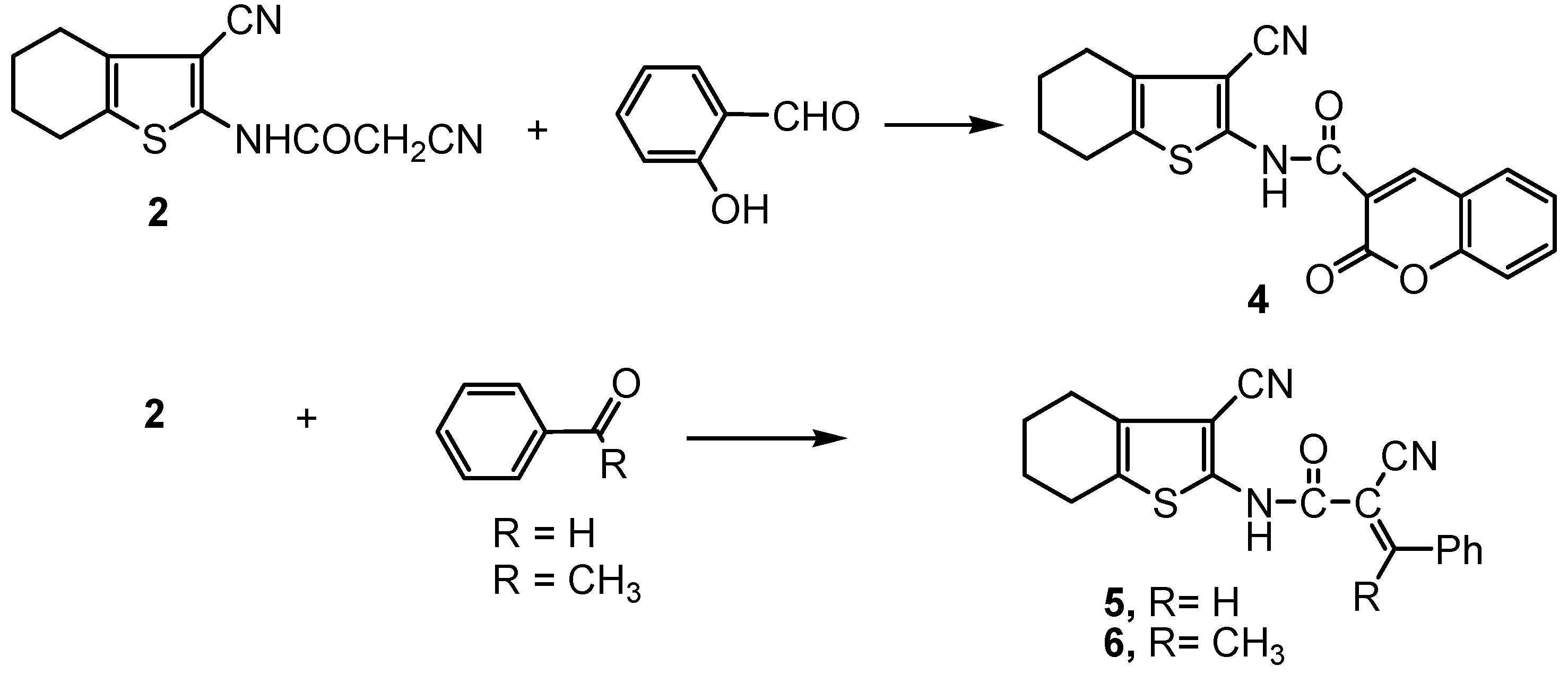
2.1. Chemistry
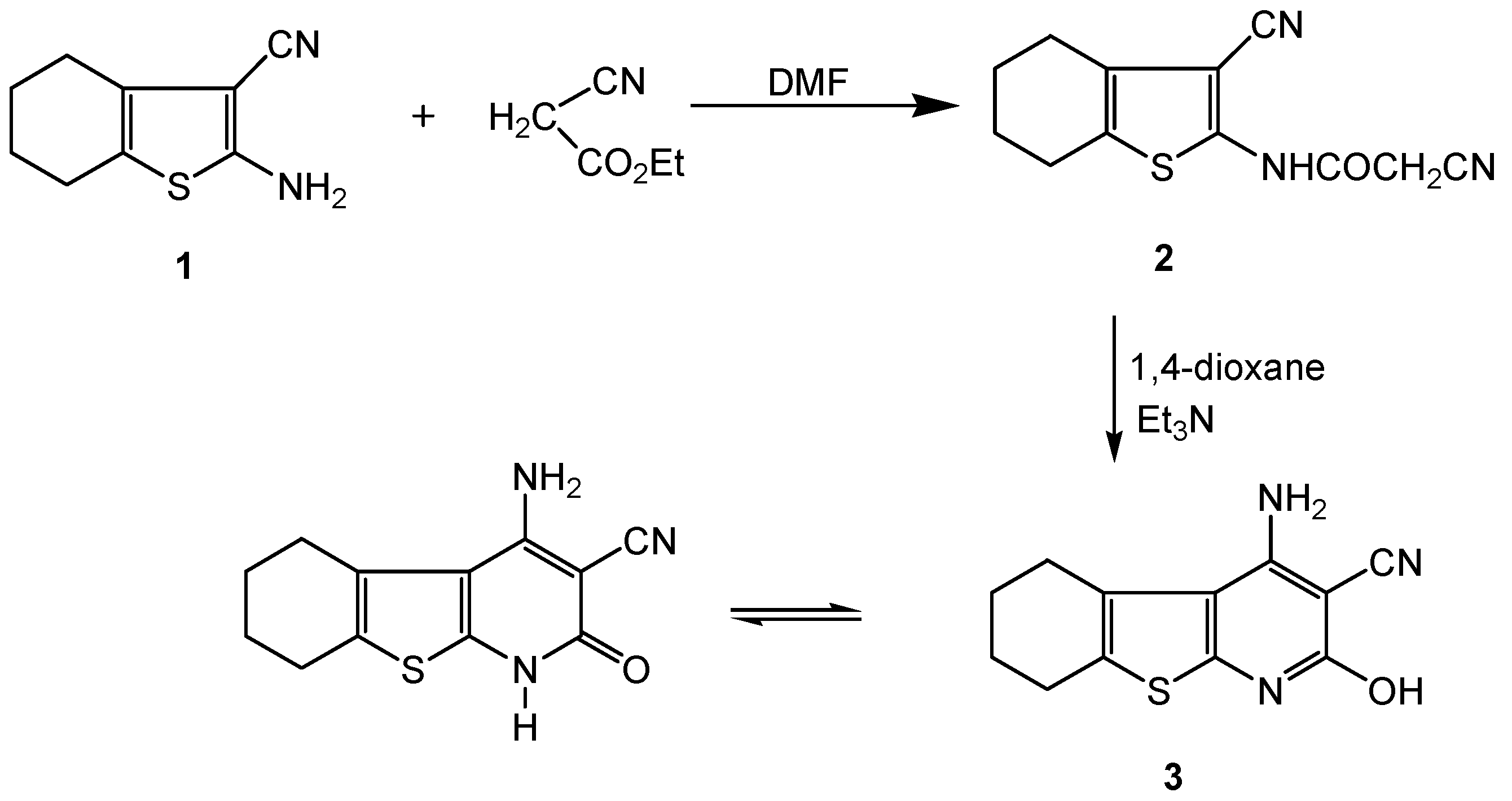
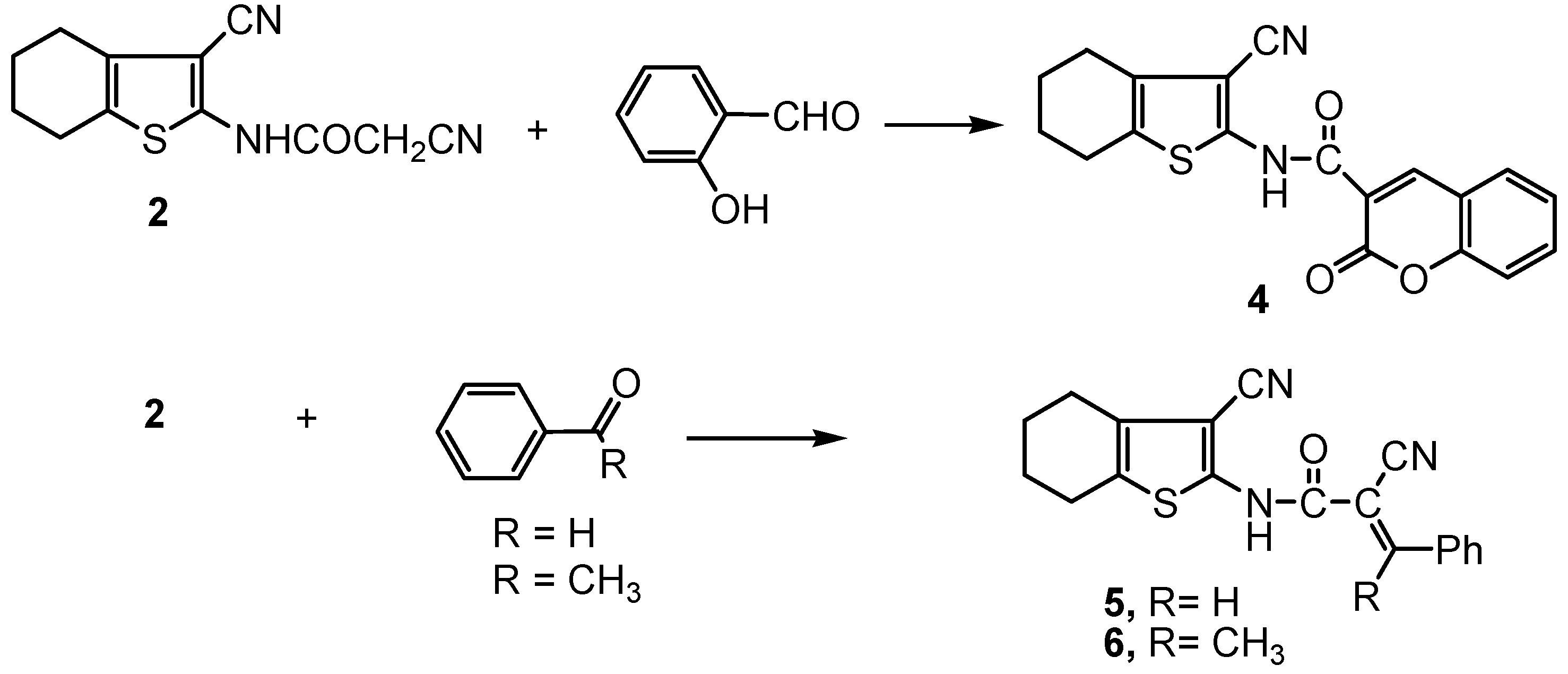
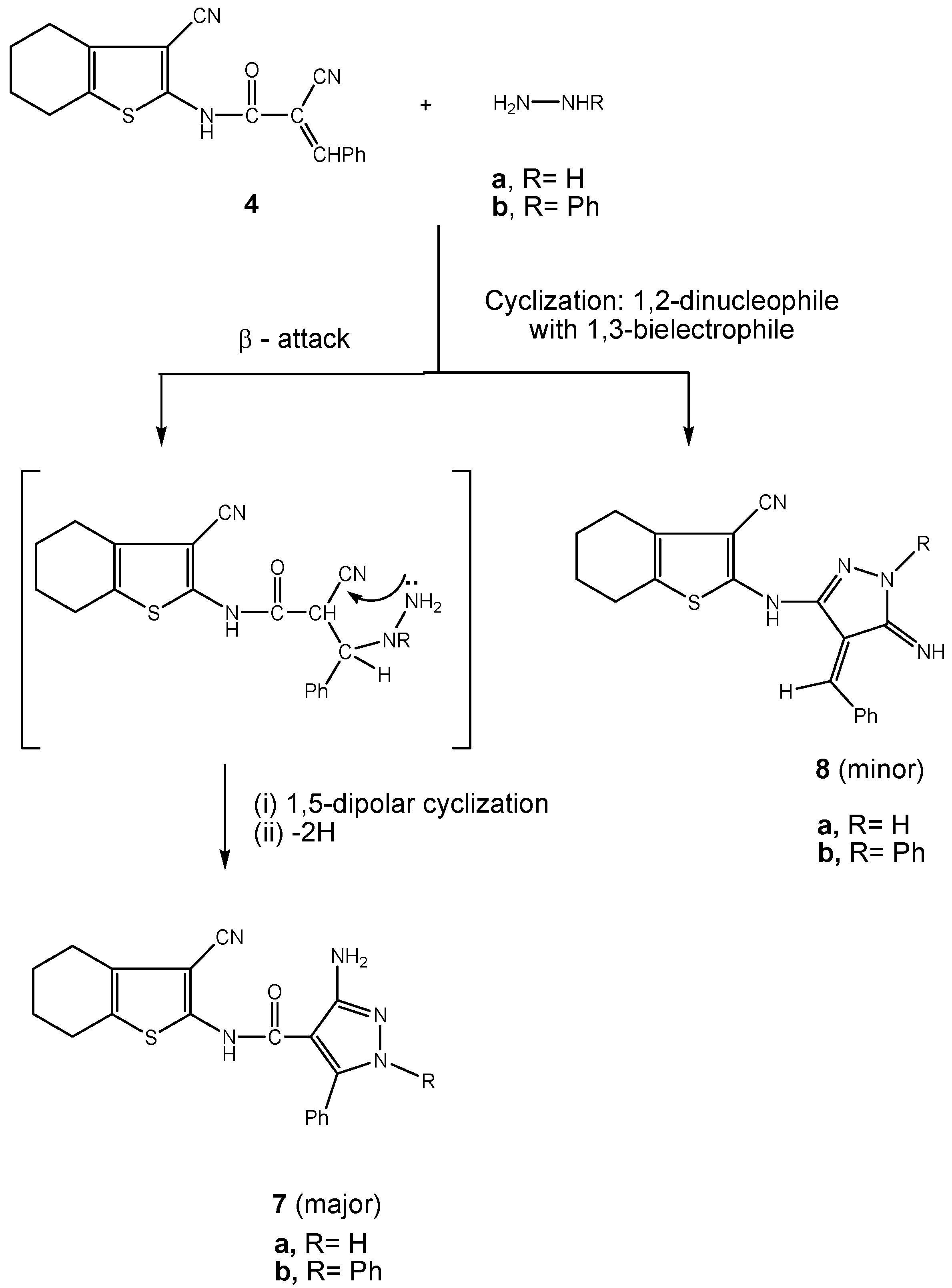
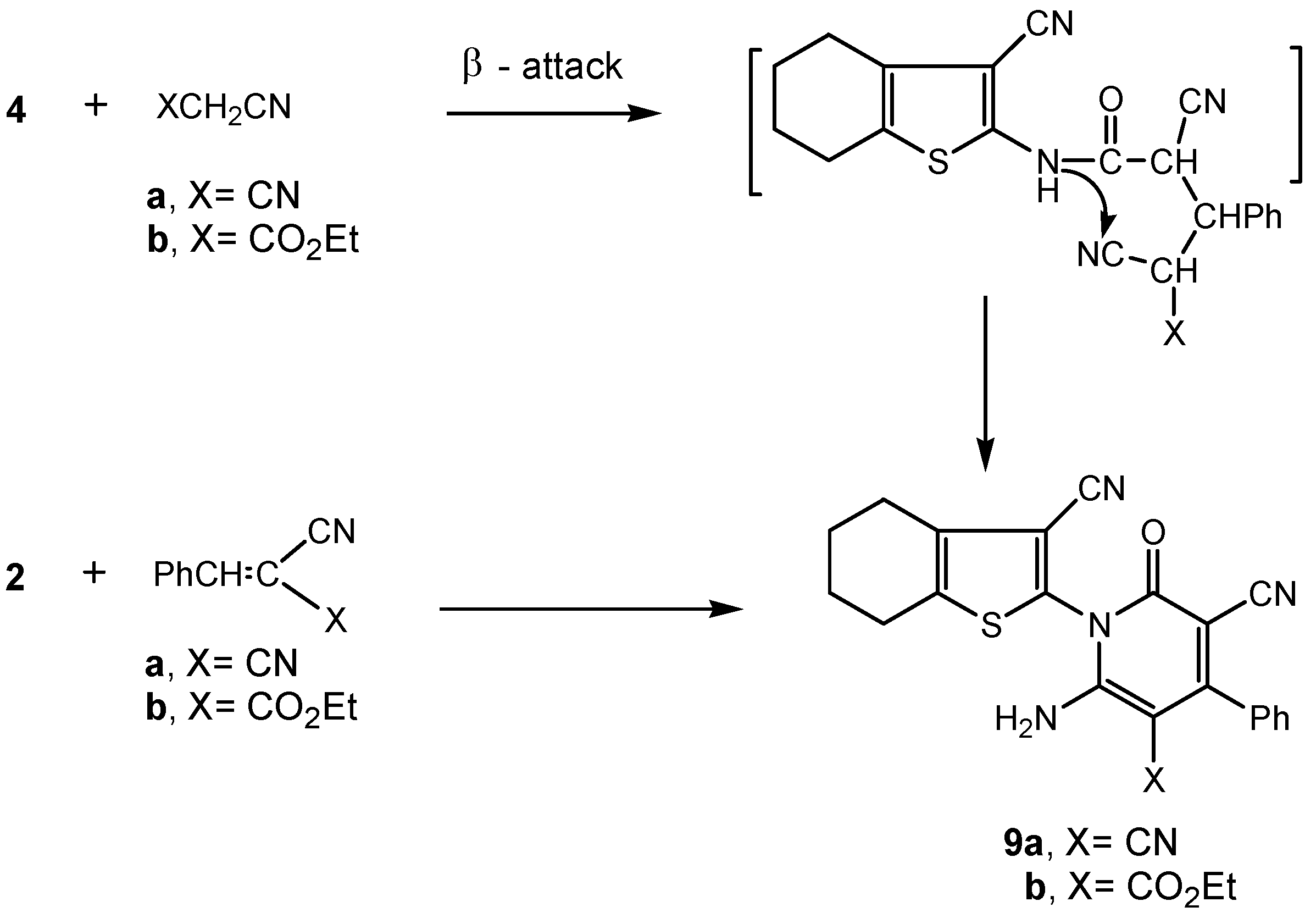
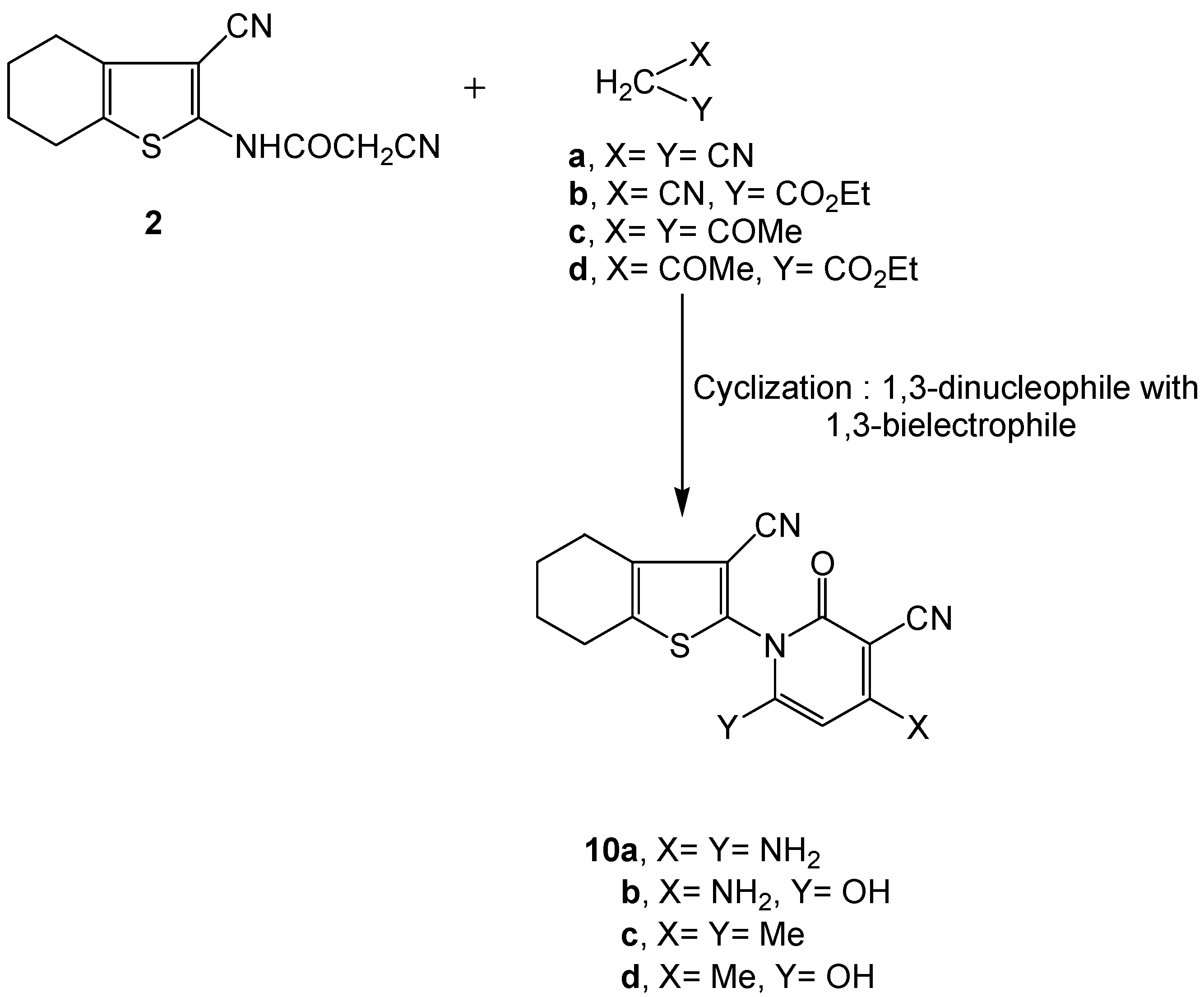
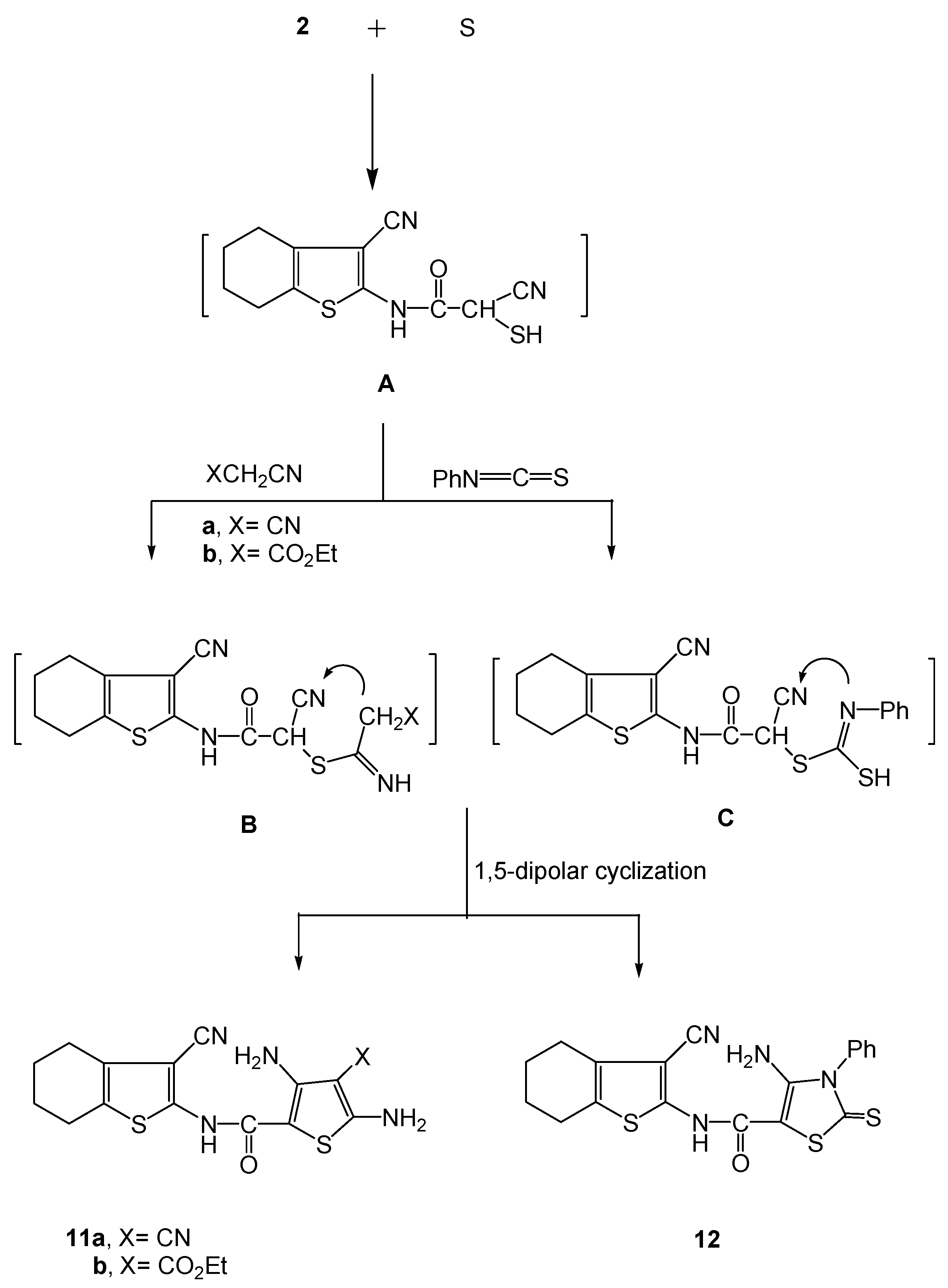

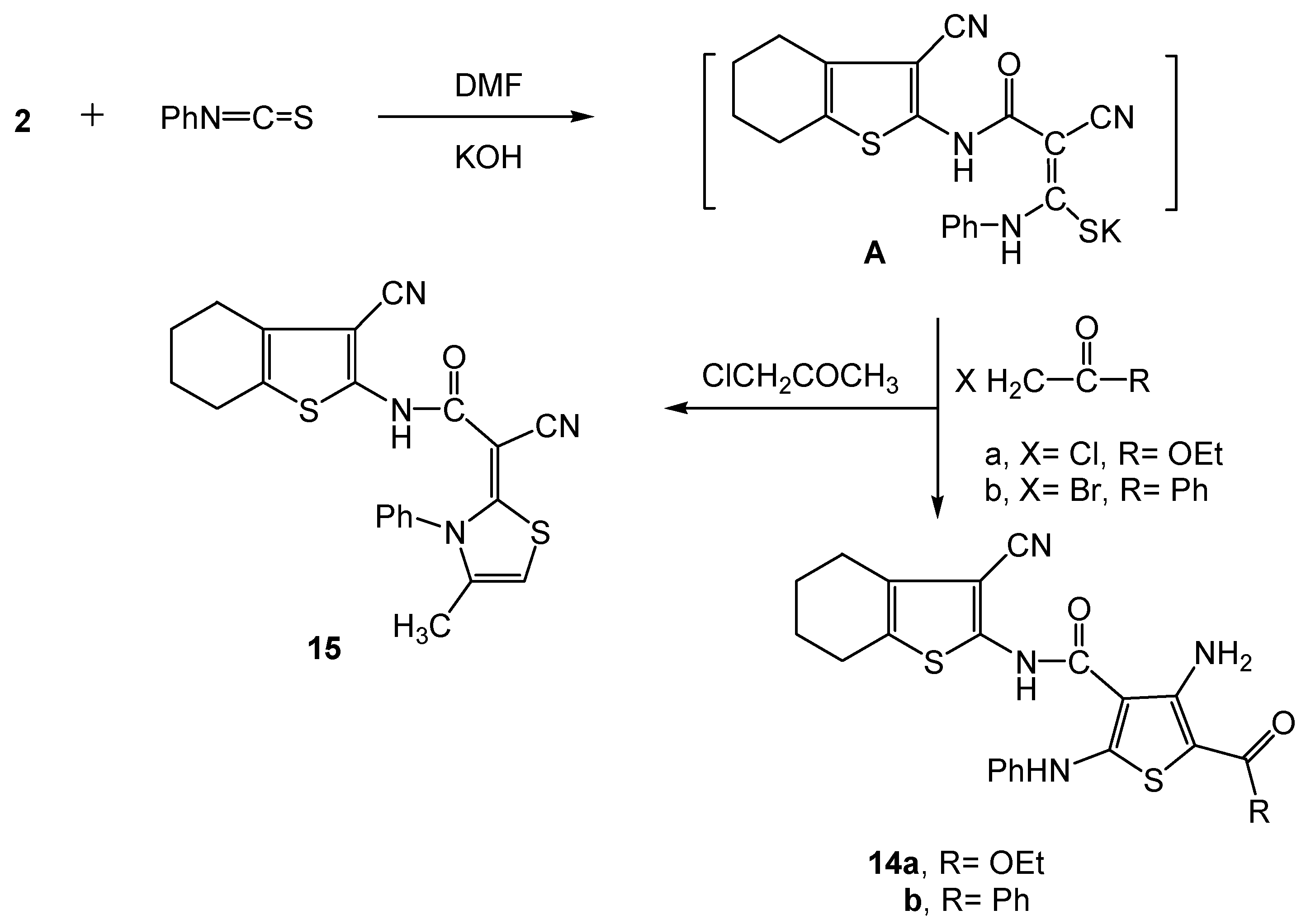
2.2. Biology
2.2.1. In vitro evaluation of antiproliferative activity of the synthesized compounds

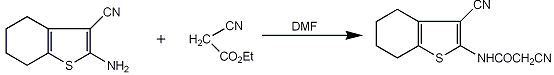{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GI50 (µM) a | ||
|---|---|---|---|
| MCF-7 | NCI-H460 | SF-268 | |
| 2 | 30.0 ± 0.6 | 19.3 ± 1.4 | 26.3 ± 1.5 |
| 3 | 44.6 ± 12.6 | 32.6 ± 8.6 | 60.4 ± 14.8 |
| 4 | 10.8 ± 0.6 | 16.5 ± 0.8 | 16.7 ± 1.6 |
| 5 | 75.7 ± 17.5 | 40.2 ± 12.8 | 52.0 ± 9.0 |
| 6 | 37.4 ± 10.2 | 22.1 ± 0.8 | 14.9 ± 6.8 |
| 7a | 2.5 ± 0.5 | 10.4 ± 0.6 | 8.0 ± 0.4 |
| 7b | 74.9 ± 0.9 | 40.6 ± 1.8 | 58.8 ± 0.8 |
| 9a | 38.0 ± 1.8 | 40.0 ± 0.8 | 22.5 ± 1.1 |
| 9b | 20.0 ± 0.2 | 30.6 ± 1.4 | 38.4 ± 0.6 |
| 10a | 16.7 ± 1.6 | 10.8 ± 0.6 | 16.5 ± 0.8 |
| 10b | 50.1 ± 0.7 | 23.2 ± 4.8 | 18.4 ± 1.8 |
| 10c | 39.0 ± 1.8 | 46.0 ± 0.8 | 22.5 ± 1.1 |
| 10d | 22.0 ± 0.2 | 30.6 ± 1.4 | 38.4 ± 0.6 |
| 11a | 66.6 ± 12.2 | 12.0 ± 6.2 | 24.8 ± 3.2 |
| 11b | 22.0 ± 0.4 | 26.3 ± 0.8 | 39.0 ± 0.8 |
| 12 | 10.9 ± 0.2 | 146.1 ± 0.6 | 22.3 ± 0.5 |
| 13 | 42.6 ± 12.2 | 32.6 ± 8.6 | 64.4 ± 14.8 |
| 14a | 20.0 ± 0.2 | 32.6 ± 1.4 | 36.4 ± 0.6 |
| 14b | 11.8 ± 0.6 | 14.5 ± 0.8 | 16.7 ± 1.6 |
| 15 | 36.4 ± 10.2 | 20.1 ± 0.8 | 18.9 ± 6.8 |
| 17a | 2.0 ± 0.4 | 8.3 ± 0.8 | 4.0 ± 0.8 |
| 17b | 68.6 ± 12.2 | 12.0 ± 6.2 | 24.8 ± 3.2 |
| *Doxorubicin | 0.0428 ± 0.0082 | 0.0940 ± 0.0087 | 0.0940 ± 0.0070 |
3. Experimental
3.1. General
3.2. Chemistry
3.2.1. 2-Cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide (2)
3.2.2. 4-Amino-2-hydroxy-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-b] pyridine-3-carbonitrile (3)
3.2.3. Synthesis of the amide derivatives 4 and 5.
3.2.4. 2-Cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-3-phenylbut-2E-enamide (6).
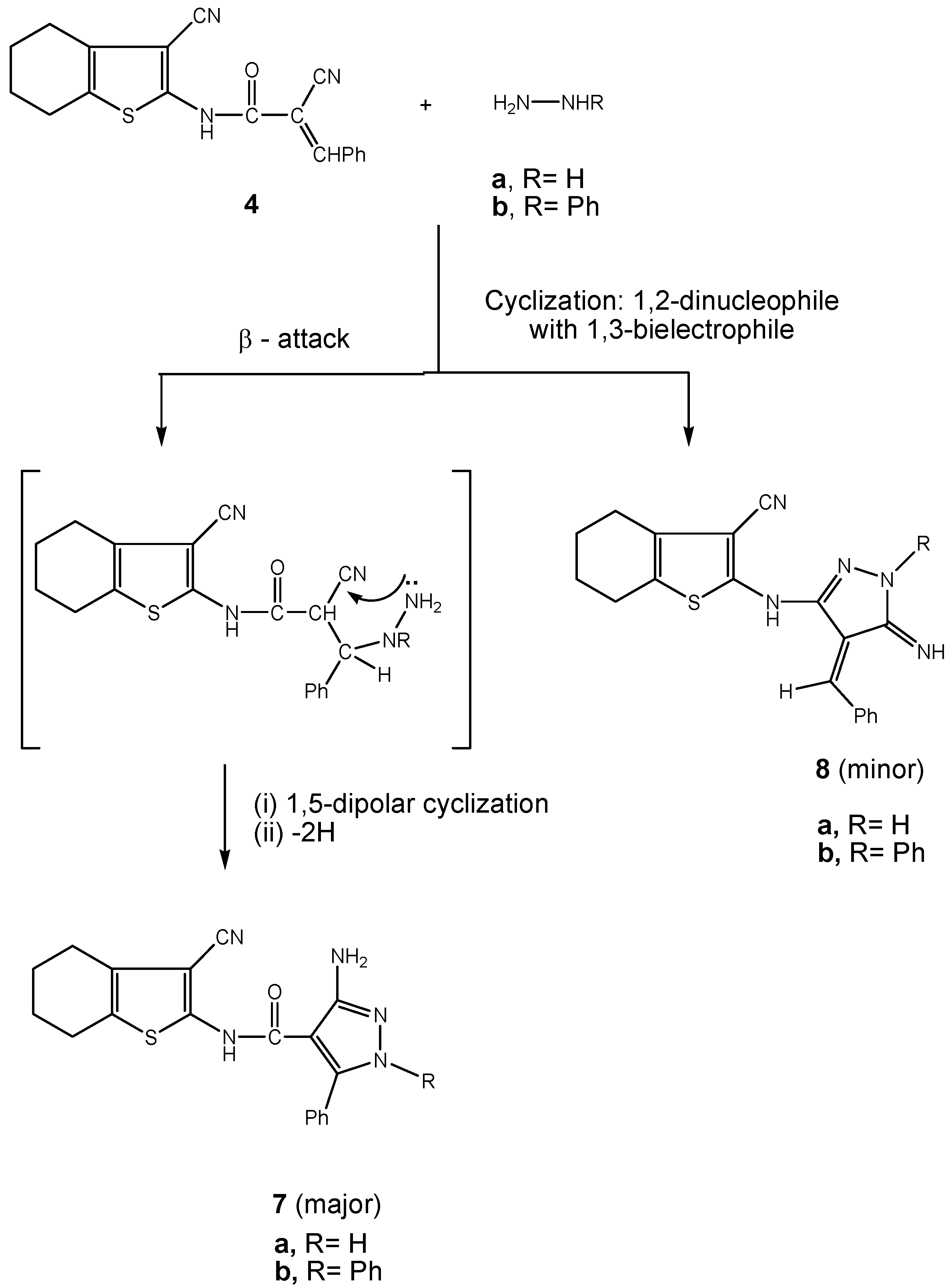
3.2.5. Synthesis of pyrazole carboxamide derivatives 7a,b
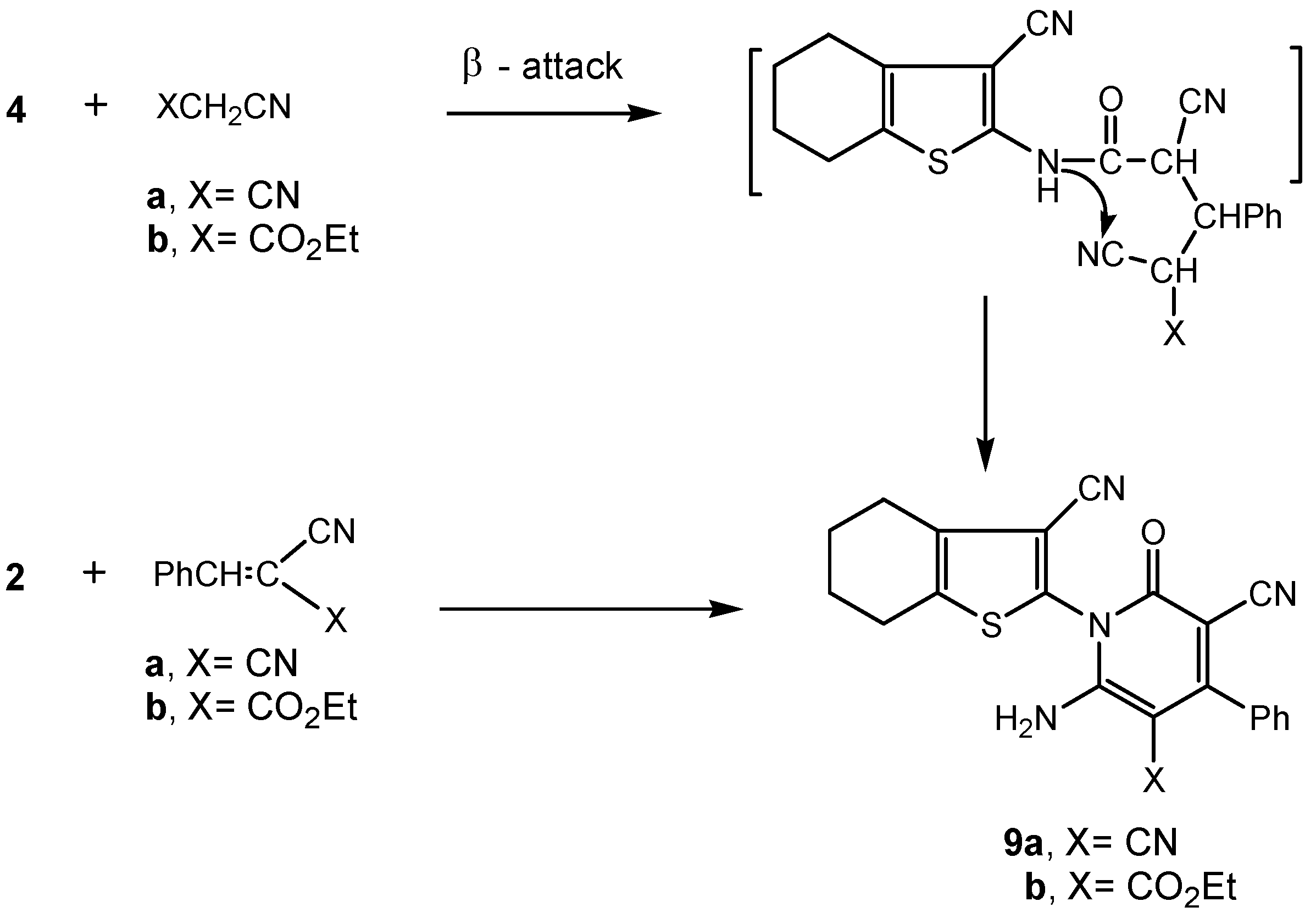
3.2.6. Synthesis of 3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl-functionalized pyridone derivatives 9a,b.
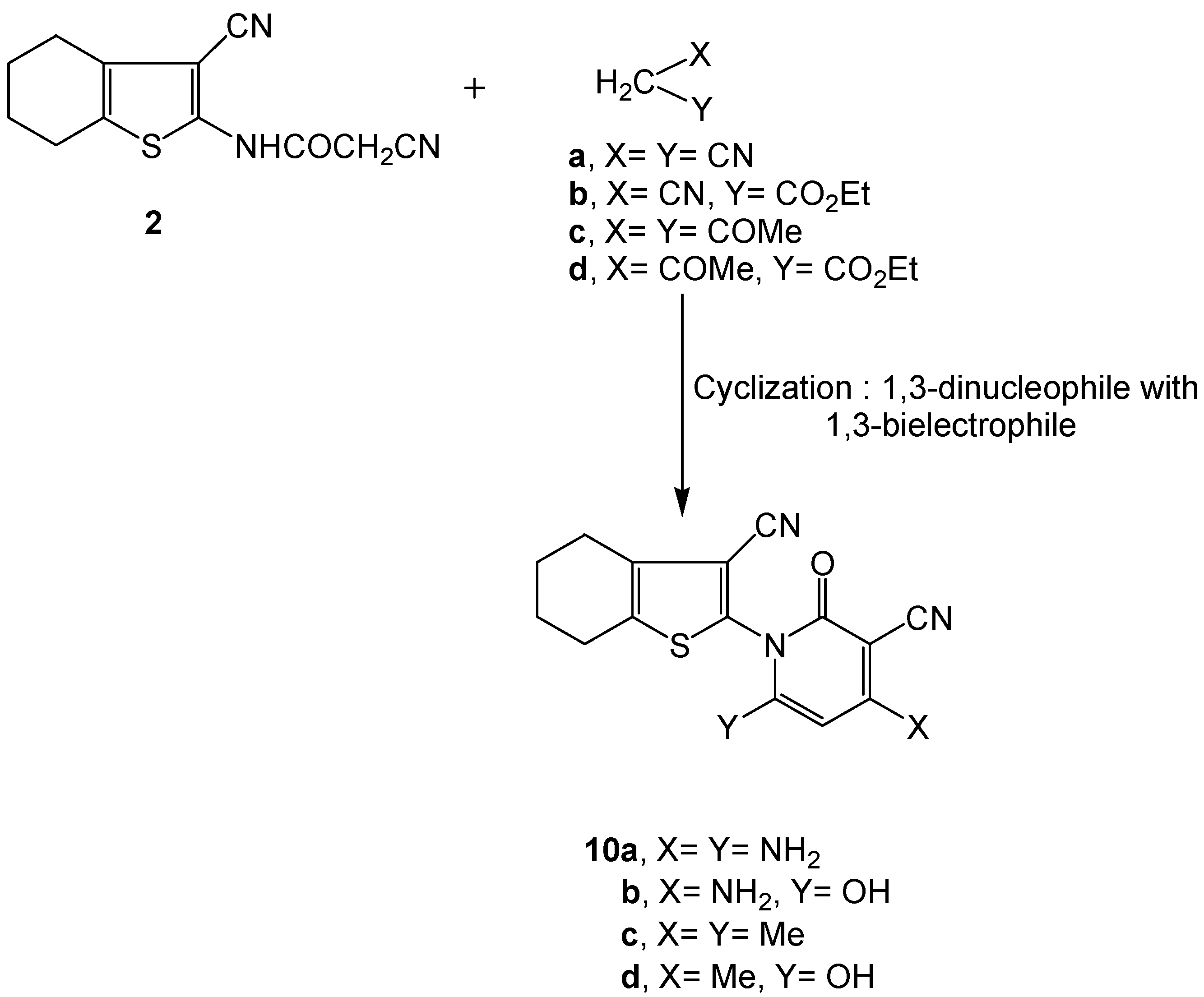
3.2.7. Synthesis of 3-cyano-4,5,6,7-tetrahydrobenzo[b]-thiophen-2-yl-functionalized 2-pyridone derivatives 10a-d.
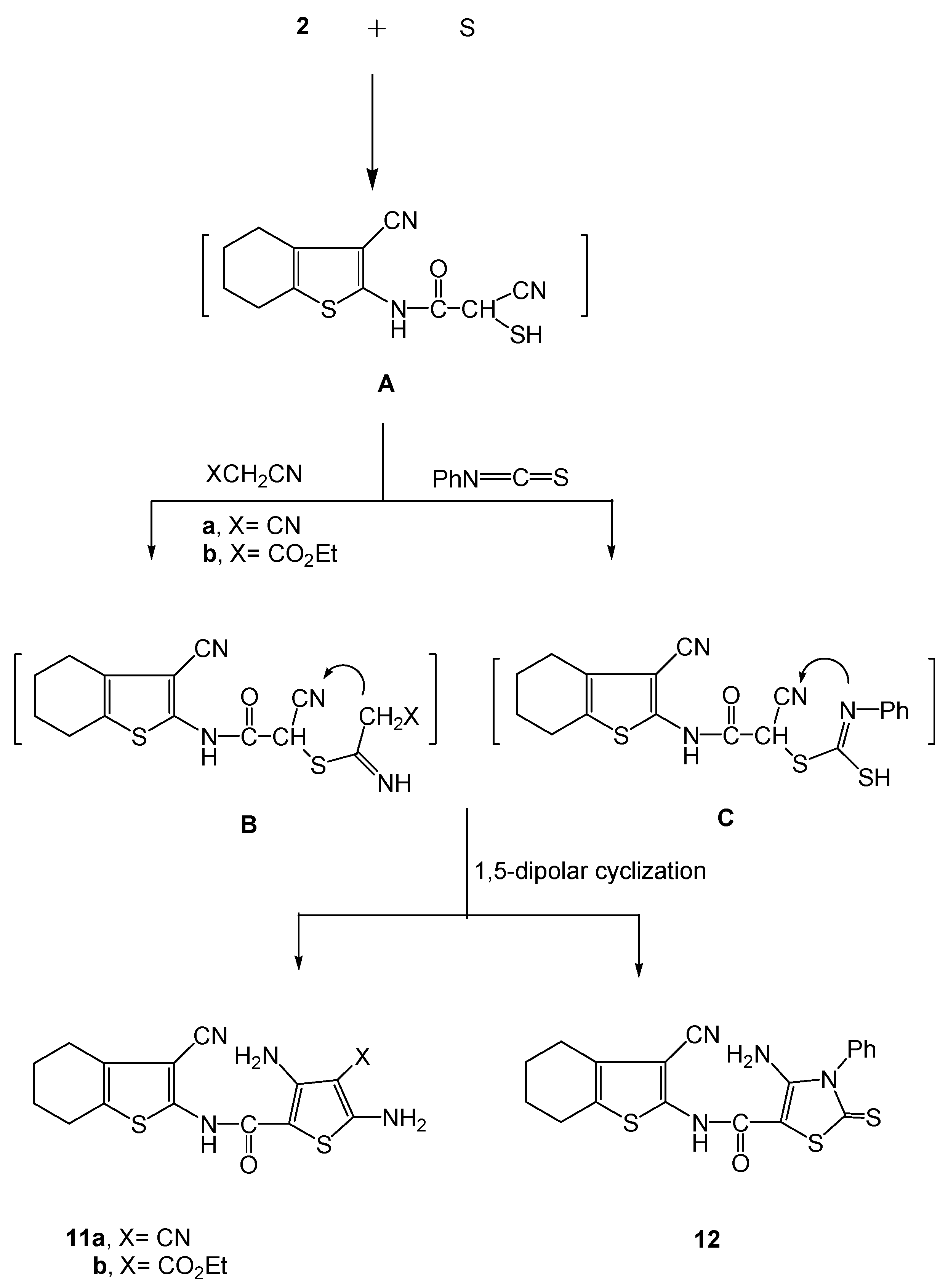
3.2.8. Synthesis of (3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)- functionalized thiophene- derivatives 11a,b and the thiazole derivative 12.
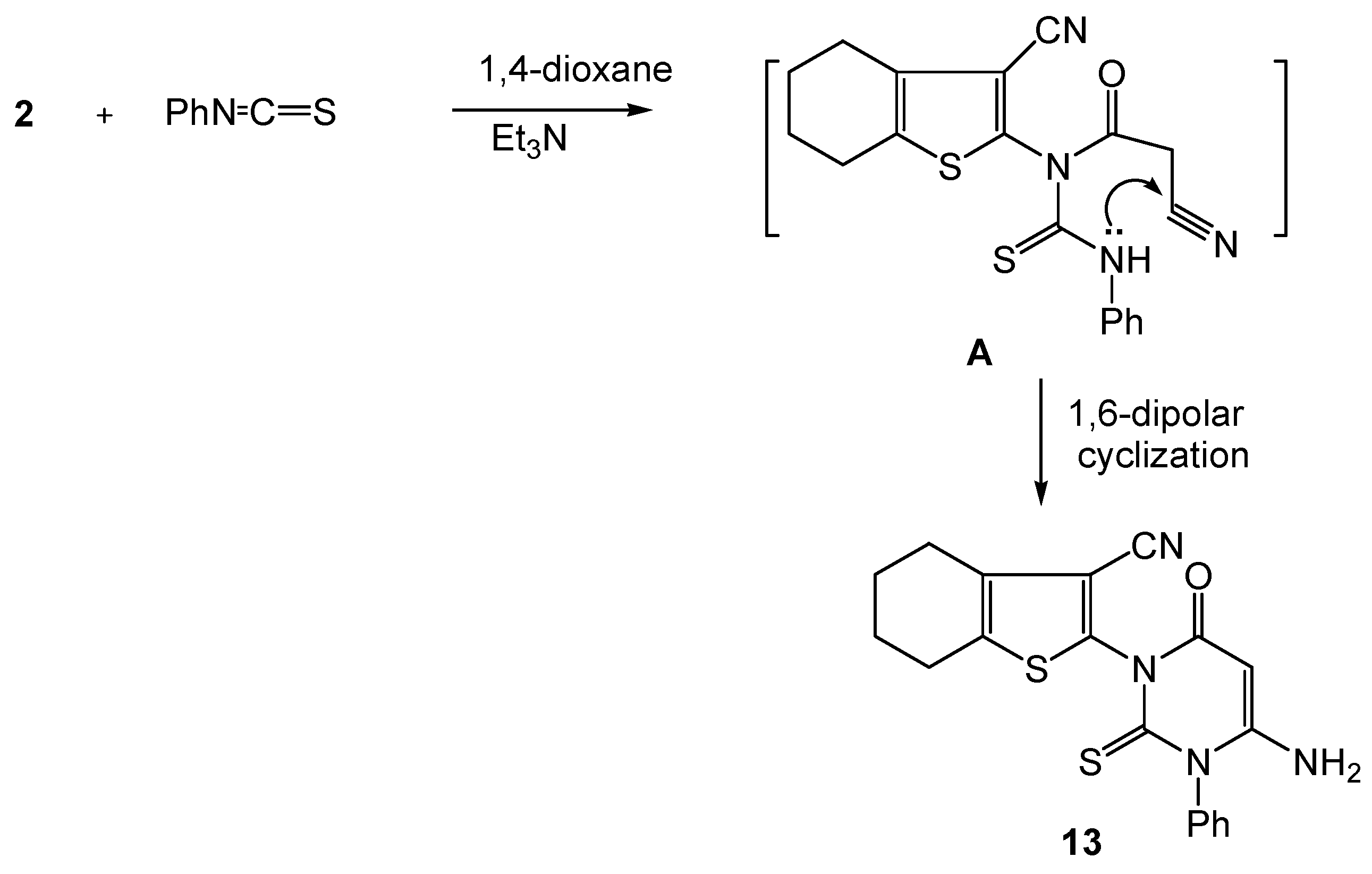
3.2.9. 2-(4-Amino-2,3-dihydro-6-oxo-3-phenyl-2-thioxopyrimidin-1(6H)-yl)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (13).
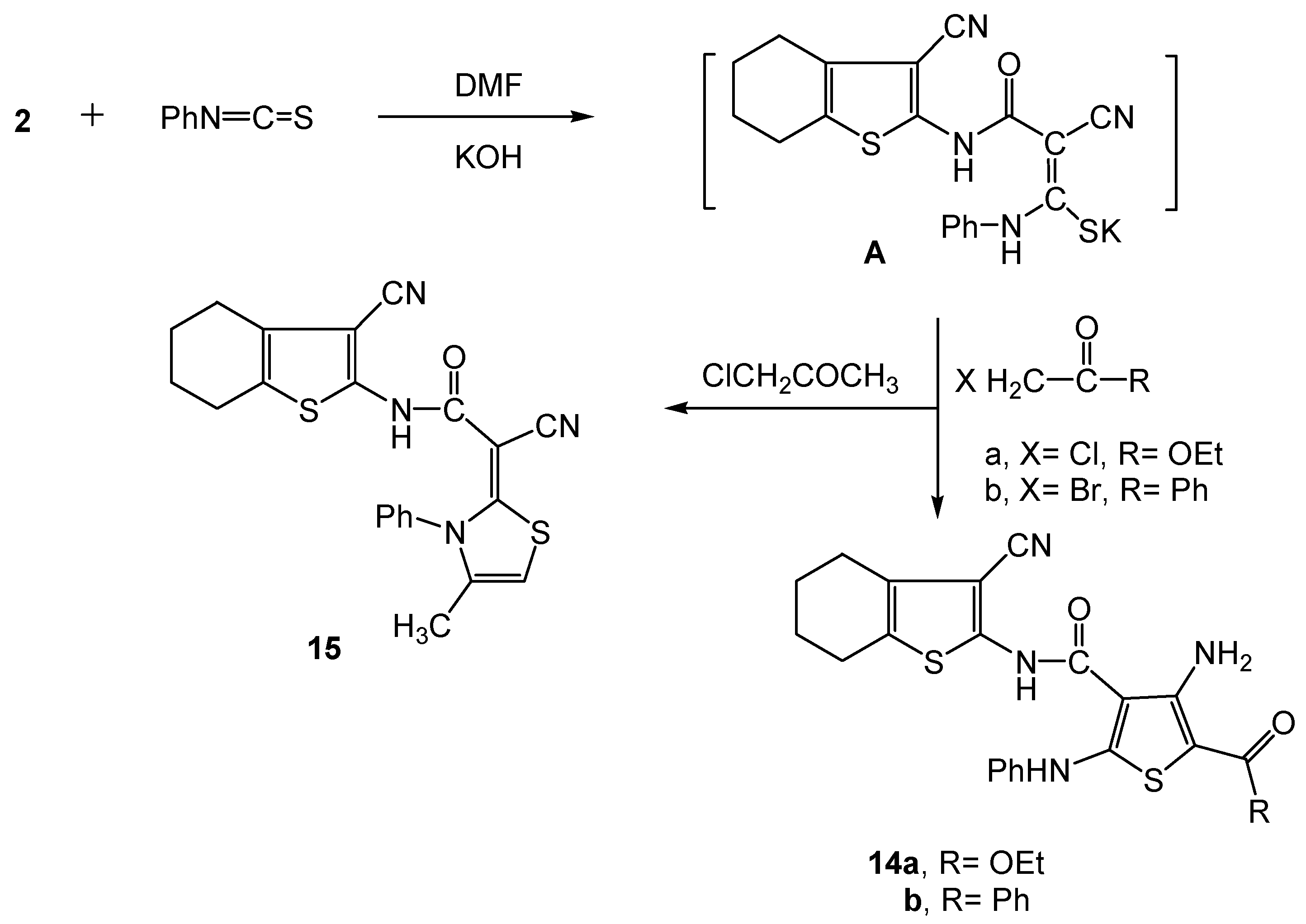
3.2.10. Synthesis of the 3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl-functionalized thiophene derivatives 14a,b and the thiazole 15 derivative.
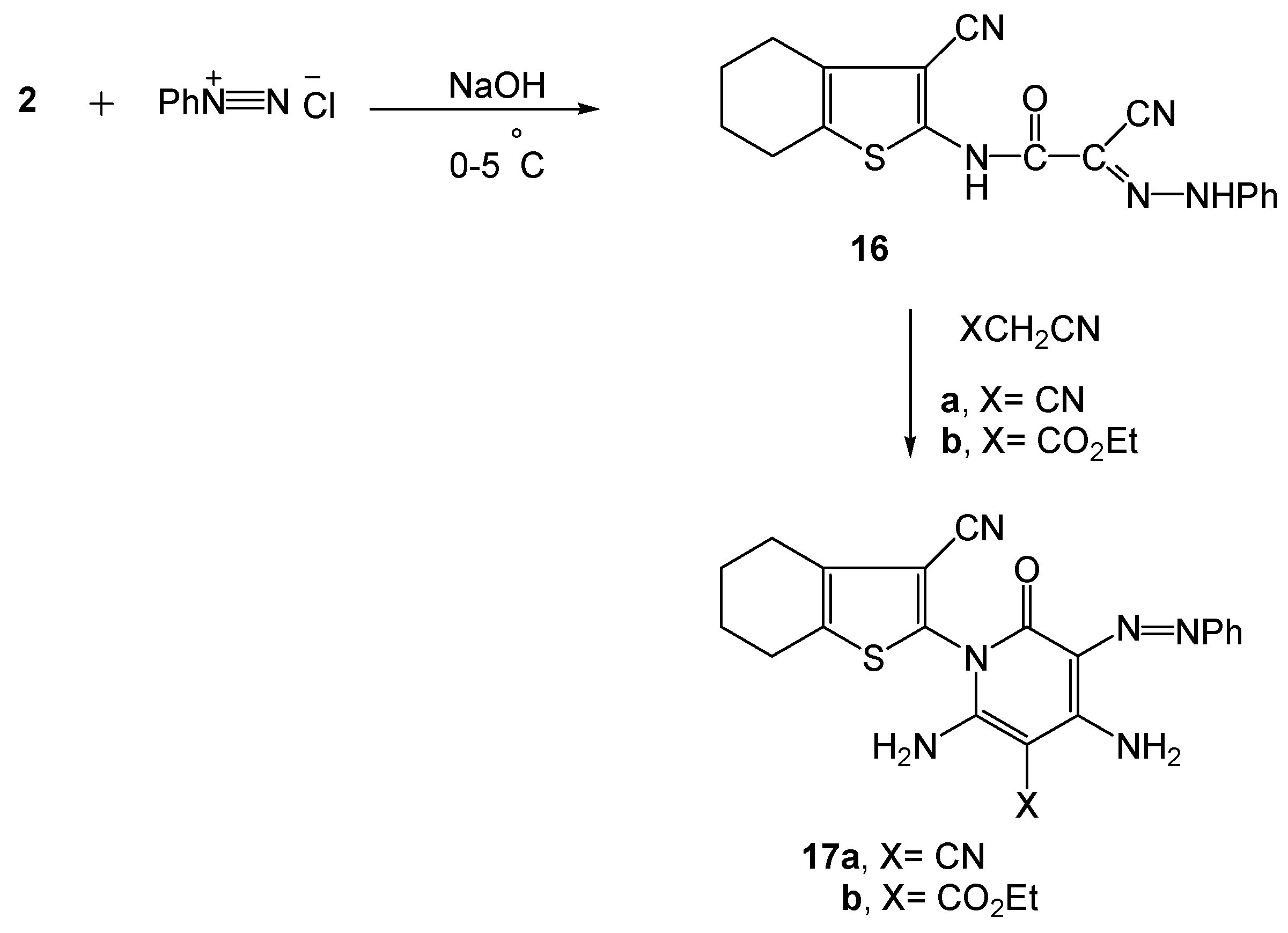
3.2.11. 2-Cyano-2-(2-phenylhydrazono)-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide (16)
3.2.12. Synthesis of 3-phenylazo-2-pyridone derivatives 17a,b
3.3. Biology
4. Conclusions
Acknowledgements
References and Notes
- Chobot, V.; Vytlačilová, J.; Kubicová, L.; Opletal, L.; Jahodář, L.; Laaskso, I.; Vuorela, P. Phototoxic activity of a thiophene polyacetylene from Leuzea carthamoides. Fitoterapia 2006, 77, 194–198. [Google Scholar] [CrossRef]
- Tor, Y.; Valle, S.D.; Jaramillo, D.; Srivatsan, S.G.; Rios, A.; Weizman, H. Designing new isomorphic fluorescent nucleobase analogues : the thieno[3,2-d]pyrimidine core. Tetrahedron 2007, 63, 3608–3614. [Google Scholar] [CrossRef]
- Okutan, M.; Yerli, Y.; San, S.E.; Yilmaz, F.; Günaydin, O.; Durak, M. Dielectric properties of thiophene based conducting polymers. Syn. Metals 2007, 157, 368–373. [Google Scholar] [CrossRef]
- Ogawa, S.; Muraoka, H.; Kikuta, K.; Saito, F.; Sato, R. Design of reversible multi-electron redox systems using benzochalcogenophenes containing aryl and/or ferrocenyl fragments. J. Organometal. Chem. 2007, 692, 60–69. [Google Scholar]
- Destri, S.; Giovanella, U.; Fazio, A.; Porzio, W.; Gabriele, B.; Zotti, G. Poly(bithiophene)-co-3,4-di(methoxycarbonyl)methyl thiophene for LED. Org. Electron. 2002, 3, 149–156. [Google Scholar] [CrossRef]
- Bouachine, M.; Benaqqa, O.; Toufik, H.; Hamidi, M.; Lère-Porte, J.-P.; Serein-Spirau, F.; Amine, A. Experimental and quantum chemical investigation of new electroluminescent material based on thiophene, phenylene and antracene. Analele Universităţii din Bucureşti 2010, 19, 35–44. [Google Scholar]
- Batista, R.M.F.; Costa, S.P.G.; Belsley, M.; Lodeiro, C.; Raposo, M.M.M. Synthesis and characterization of novel (oligo)thienyl-imidazo-phenanthrolines as versatile π conjugated systems for several optical applications. Tetrahedron 2008, 64, 9230–9238. [Google Scholar]
- Jarvest, R.L.; Pinto, I.L.; Ashman, S.M.; Dabrowski, C.E.; Fernandez, A.V.; Jennings, L.J.; Lavery, P.; Tew, D.G. Inhibition of herpes proteases and antiviral activity of 2-substituted thieno[2,3-d]oxazinones. Bioorg. Med. Chem. Lett. 1999, 9, 443–448. [Google Scholar] [CrossRef]
- Wardakhan, W.W.; Shams., H.Z.; Moustafa, H.E. Synthesis of polyfunctionally substituted thiophene, thieno [2,3-b]pyridine and thieno[2,3-d]pyrimidine derivatives. Phosph. Sulf. Silicon 2005, 180, 1815–1827. [Google Scholar] [CrossRef]
- Isabel, C.F.R.; Ricardo, C. C.; Letícia, M. E.; Maria-João, R.P.Q. Screening of antimicrobial activity of diarykamines in the 2,3,5-trimethylbenzo[b]thiopheneseries: a structure-activity evaluation study. Bioorg. Med. Chem. Lett. 2004, 14, 5831–5833. [Google Scholar] [CrossRef]
- Pinto, E.; Queiroz, M.-J.R.P.; Vale-Silva, L.A.; Oliveira, J.F.; Begouin, A.; Begouin, J.-M.; Kirsch, G. Antifungal activity of synthetic di(hetero)arylamines based on the benzo[b]thiophene moiety. Bioorg. Med. Chem. 2008, 16, 8172–8177. [Google Scholar] [CrossRef]
- Dannhardt, G.; Kiefer, W.; Krämer, G.; Maehrlein, S.; Nowe, U.; Fiebich, B. The pyrrole moiety as a template for COX-1/COX-2 inhibitors. Eur. J. Med. Chem. 2000, 35, 499–510. [Google Scholar] [CrossRef]
- Brault, L.; Migianu, E.; Néguesque, A.; Battaglia, E.; Bagrel, D.; Kirsch, G. New thiophene analogues of kenpaullone: synthesis and biological evaluation in breast cancer cells. Eur. J. Med. Chem. 2005, 40, 757–763. [Google Scholar] [CrossRef]
- Starčević, K.; Karminski-Zamola, G.; Piantanida, I.; Zinić, M.; Sǔman, L.; Kralj, M. Photoinduced switch of a DNA/RNA inactive molecule into a classical intercalator. J. Am. Chem. Soc. 2005, 127, 1074–1075. [Google Scholar]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Preti, D.; Tolomeo, M.; Grimaudo, S.; Cristina, A.D.; Zonta, N.; Balzarini, J.; Brancale, A.; Sarkar, T.; Hamel, E. Design, synthesis and biological evaluation of thiophene analogues of chalcones. Bioorg. Med. Chem. 2008, 16, 5367–5376. [Google Scholar]
- Ferreira, I.C.F.R.; Queiroz, M.R.P.; Vilas-Boas, M.; Estevinho, L. M.; Begouin, A.; Kirsch, G. Evaluation of the antioxidant properties of diarylamines in benzo[b]thiophene series by free radical scavenging activity and reducing power, Bioorg. Med. Chem. Lett. 2006, 16, 1384–1387. [Google Scholar] [CrossRef]
- Pinto, I.L.; Jarvest, R.L.; serafinowska, H.T. The synthesis of 5-alkoxy and 5-amino substituted thiophenes. Tetrahedron Lett. 2000, 41, 1597–1600. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Pavani, M.G.; Shryock, J.C.; Moorman, A.R.; Iannotta, V.; Borea, P.A.; Romagnoli, R. Synthesis of 2-amino-3-heteroaroylthiophenes and evaluation of their activity as potential allosteric enhancers at the human A1 receptor. Eur. J. Med. Chem. 2004, 39, 855–865. [Google Scholar] [CrossRef]
- Pillai, A.D.; Rathod, P.D.; Xavier, F.P.; Vasu, K.K.; Padh, H.; Sudarsanam, V. Design, synthesis, and pharmacological evaluation of some 2-[4-morpholino]-3-aryl-5-substituted thiophenes as novel anti-inflammatory agents: generation of a novel anti-inflammatory pharmacophore. Bioorg. Med. Chem. 2004, 12, 4667–4671. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Zali-Boinee, H. A versatile one-pot synthesis of 2,3,5-tri-substituted thiophenes from thiomorpholides. Tetrahedron Lett. 2003, 44, 6253–6255. [Google Scholar] [CrossRef]
- Jones, R.J.; Cava, M.P. Photocyclisation strategy for the synthesis of antitumour agent CC-1065: synthesis of thiophene analogues of PDE-I and PDE-II. J. Chem. Soc. Chem. Commun. 1986, 826–827. [Google Scholar] [CrossRef]
- Starčević, K.; Kralj, M.; Piantanida, I.; Šuman, L.; Pavelić, K.; Karminski-Zamola, G. Synthesis, photochemical synthesis, DNA binding and antitumor evaluation of novel cyano- and amidino-substituted derivatives of naphtho-furans, naphtho-thiophenes, thieno-benzofurans, benzo-dithiophenes and their acyclic precursors. Eur. J. Med. Chem. 2006, 41, 925–939. [Google Scholar] [CrossRef]
- James, B.; Suresh, E.; Nair, M.S. Friedel-Crafts alkylation of a cage enone: synthesis of aralkyl substituted tetracyclo[5.3.1.0[2,6].0[4,8]]undeca-9,11-diones and the formation of fascinating novel cage compounds with pyrrole and thiophene using Montmorillonite K-10. Tetrahedron Lett. 2007, 48, 6059–6063. [Google Scholar] [CrossRef]
- Kondolff, I.; Doucet, H.; Santelli, M. Synthesis of biheteroaryl derivatives by tetraphosphine/palladium-catalysed Suzuki coupling of heteroaryl bromides with heteroarylboronic acids. J. Mol. Catal. A: Chem. 2007, 269, 110–118. [Google Scholar] [CrossRef]
- Queiroz, M.-J.R.P.; Ferreira, I.C.F.R.; Gaetano, Y.D.; Kirsch, G.; Calhelha, R.C.; Estevinho, L.M. Synthesis and antimicrobial activity studies of orthochlorodiarylamines and heteroaromatic tetracyclic systems in the benzo[b]thiophene series. Bioorg. Med. Chem. 2006, 14, 6827–6831. [Google Scholar] [CrossRef]
- Quèiroz, M.-J.R.P.; Ferreira, I.C.F.R.; Calhelha, R.C.; Estevinho, L.M. Synthesis and antioxidant activity evaluation of new 7-aryl or 7-heteroarylamino-2,3-dimethylbenzo[b]thiophenes obtained by Buchwald-Hartwig C-N cross-coupling. Bioorg. Med. Chem. 2007, 15, 1788–1794. [Google Scholar] [CrossRef]
- Rodriguez, J.G.; Esquivias, J.; Lafuente, A.; Rubio, L. Synthesis of conjugated 2 and 2,5-(ethenyl) and (ethynyl)phenylethynyl thiophenes: fluorescence properties. Tetrahedron 2006, 62, 3112–3122. [Google Scholar] [CrossRef]
- Raju, S.; Kumar, P.R.; Mukkanti, K.; Annamalai, P.; Pal, M. Facile synthesis of substituted thiophenes via Pd/C-mediated sonogashira coupling in water. Bioorg. Med. Chem. Lett. 2006, 16, 6185–6189. [Google Scholar] [CrossRef]
- Shams, H.Z.; Elkholy, Y.M.; Azzam, R.A.; Mohareb, R.M. Synthetic potentialities of thiophene systems in heterocyclic synthesis: Anovel synthesis of thieno[2,3-b]pyridine derivatives. Phosph. Sulf. Silicon 1999, 155, 215–233. [Google Scholar] [CrossRef]
- Shams, H.Z.; Mohareb, R.M.; Helal, M.H.; Mahmoud, A.E. Synthesis, structure elucidation, and biological evaluation of some fused and/or pendant thiophene, pyrazole, imidazole, thiazole, triazole, triazine, and coumarin systems based on cyanoacetic 2-[(benzoylamino)thioxomethyl]-hydrazide. Phosph. Sulf. Silicon 2007, 182, 237–263. [Google Scholar] [CrossRef]
- Sherif, S.M.; Mohareb, R.M.; Shams, H.Z.; Gaber, H.M. A convenient synthesis of polyfunctionally substituted benzo[b]thiophen-2-yl-pyrimidine,-pyrazole,-isoxazole and-pyridazine derivatives. J. Chem. Res. 1995, (S), 434–435, (M) 2658-2674. [Google Scholar]
- Gewald, K.; Schindler, R. Cyclisierungen mit Cyanthioacetamid in Gegenwart von Schwefel. J. Prakt. Chem. 1990, 332, 223–228. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Aziz, S.I.; Sayed, N.I.A.; Shams, H.Z. Reactions of benzoyl isothiocyanate with active methylene reagents: A novel synthesis of thiophene, thiazoline and thieno[2,3-d]pyrimidine derivatives. J. Chin. Chem. Soc. 1992, 39, 181–187. [Google Scholar]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; Gray-Goodrich, M.; Campbell, H.; Mayo, J.; Boyd, M. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. NCI. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Li, L.; Yu, F.-H. Transcriptional specificities of adriamycin. Biochem. Mol. Biol. Int. 1993, 31, 879–887. [Google Scholar]
- Sample Availability: Samples of the compounds 2-17b are available from the authors.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shams, H.Z.; Mohareb, R.M.; Helal, M.H.; Mahmoud, A.E. Novel Synthesis and Antitumor Evaluation of Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-Cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide. Molecules 2011, 16, 52-73. https://doi.org/10.3390/molecules16010052
Shams HZ, Mohareb RM, Helal MH, Mahmoud AE. Novel Synthesis and Antitumor Evaluation of Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-Cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide. Molecules. 2011; 16(1):52-73. https://doi.org/10.3390/molecules16010052
Chicago/Turabian StyleShams, Hoda Z., Rafat M. Mohareb, Maher H. Helal, and Amira E. Mahmoud. 2011. "Novel Synthesis and Antitumor Evaluation of Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-Cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide" Molecules 16, no. 1: 52-73. https://doi.org/10.3390/molecules16010052
APA StyleShams, H. Z., Mohareb, R. M., Helal, M. H., & Mahmoud, A. E. (2011). Novel Synthesis and Antitumor Evaluation of Polyfunctionally Substituted Heterocyclic Compounds Derived from 2-Cyano-N-(3-cyano-4,5,6,7-tetrahydrobenzo[b]thiophen-2-yl)-acetamide. Molecules, 16(1), 52-73. https://doi.org/10.3390/molecules16010052