Convenient Synthesis of 3,4-Dichloro-5-hydroxy-2(5H)-Furanone Glycoconjugates

Abstract
:1. Introduction
2. Results and Discussion


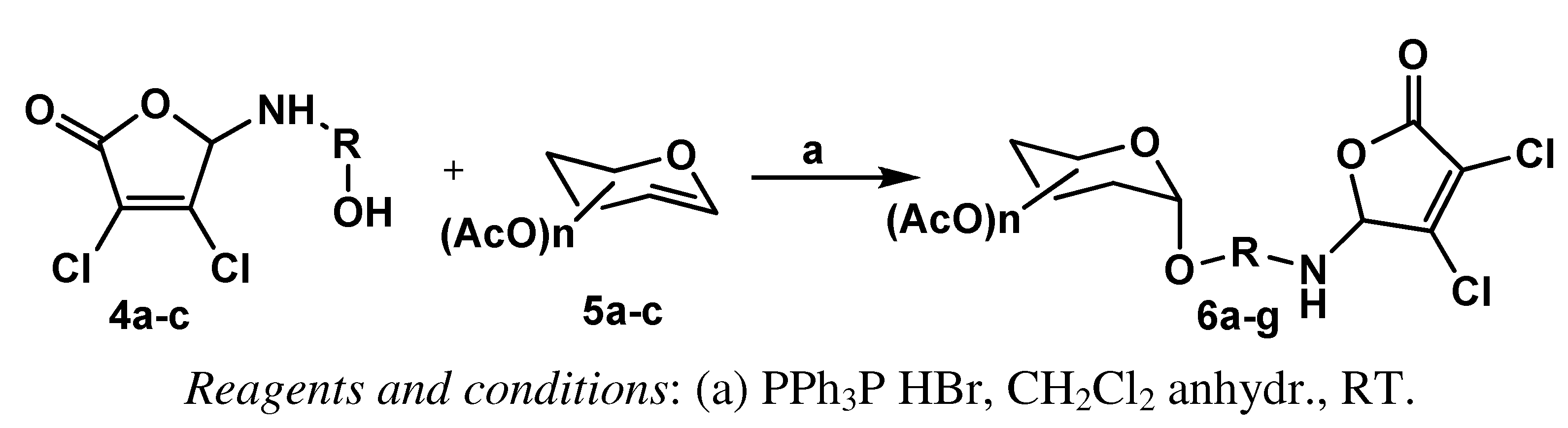

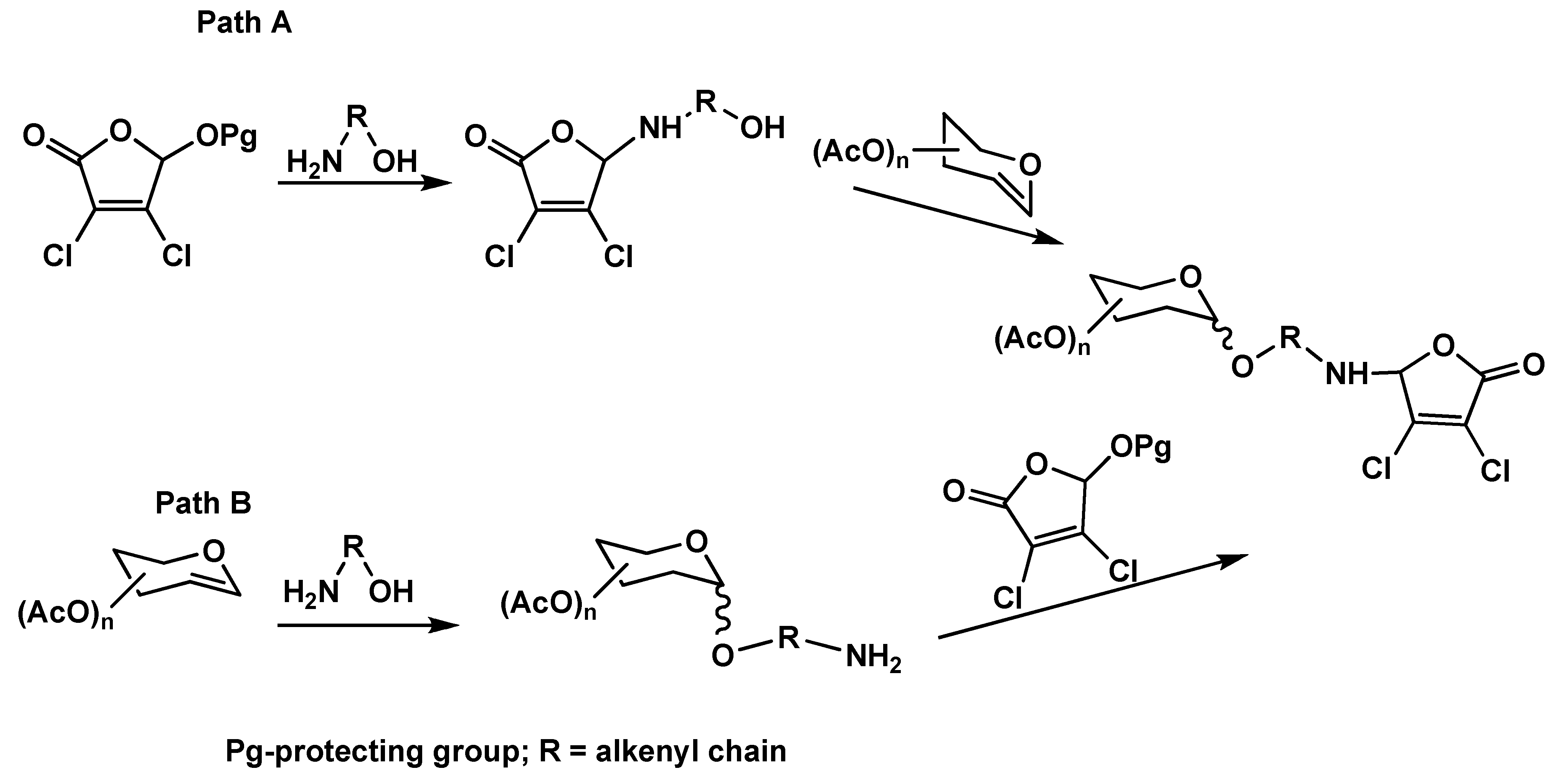{kind=link}
{kind=link}
{kind=link}
| Entry | Conjugate | Peracetylated Glucal | MCA Derivative | Yield [%] |
| 1 | 6a | D-Glucal | 4a | 98 |
| 2 | 6b | 4b | 78 | |
| 3 | 6c | 4c | 78 | |
| 4 | 6d | D-Galactal | 4a | 89 |
| 5 | 6e | 4b | 80 | |
| 6 | 6f | L-Rhamnal | 4a | 78 |
| 7 | 6g | 4b | 60 | |
| 8 | 6h | 4c | 61 |
3. Experimental
3.1. General
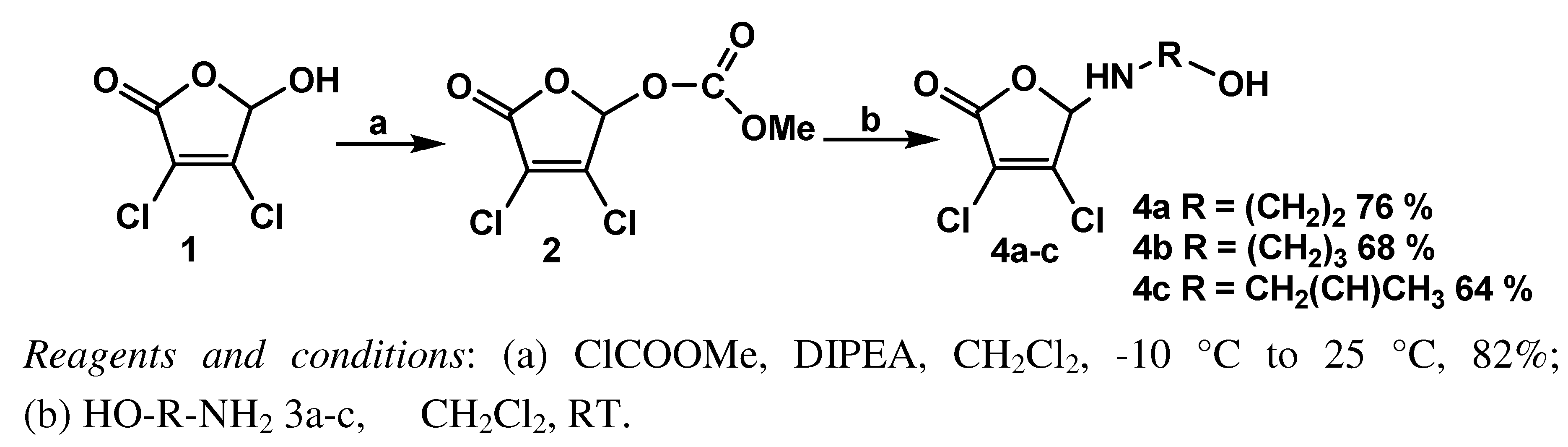
3.2. General procedure for the synthesis of 3,4-dichloro-5-( ω-hydroxyalkylamino)-2(5H)-furanones 4
3.2. General procedure for the synthesis of compounds 6a-g
4. Conclusions
Acknowledgements
References and Notes
- Bellina, F.; Rossi, R. Mucochloric and mucobromic acids: inexpensive, highly functionalised starting materials for the selective synthesis of variously substituted 2(5H)-furanone derivatives, sulfur- or nitrogen-containing heterocycles and stereodefined acyclic unsaturated dihalogenated compounds. Curr. Org. Chem. 2004, 8, 1089–1103. [Google Scholar] [CrossRef]
- Bellina, F.; Anselmi, Ch.; Martina, F.; Rossi, R. Mucochloric acid: a useful synthon for the selective synthesis of 4-aryl-3-chloro-2(5H)-furanones, (Z)-4-aryl-5-[1-(aryl)methylidene]-3-chloro-2(5H)-furanones and 3,4-diaryl-2(5H)-furanones. Eur. J. Org. Chem. 2003, 2290–2302. [Google Scholar]
- Sulikowski, G. A.; Agnelli, F.; Corbett, R. M. Investigation into a biomimetic approach toward CP-225,917 and CP-263,114. J. Org. Chem. 2000, 65, 337–342. [Google Scholar] [CrossRef]
- Angell, P.; Zhang, J.; Belmont, D. T.; Davidson, J. G. Mucohalic acid in Lewis catalyzed Mukaiyama aldol reaction: a concise method for highly functionalized γ-substituted γ-butenolides. Tetrahedron Lett. 2005, 46, 2029–2032. [Google Scholar] [CrossRef]
- Lattmann, E.; Kinchington, D.; Dunn, S.; Singh, H.; Ayuko, W. O.; Tisdale, M. J. Cytotoxicity of 3,4-dihalogenated 2(5H)-furanones. J. Pharm. Pharmacol. 2004, 56, 1163–1170. [Google Scholar]
- Mowry, D.T. Mucochloric acid. I. Reactions of the pseudo acid group. J. Am. Chem. Soc 1950, 72, 2535–2537. [Google Scholar] [CrossRef]
- Blazecka, P.G.; Belmont, D.; Curran, T.; Pflum, D.; Zhang, J. Further utilizaton of mucohalic acids: Palladium-free, regioselective etherification and amination of α,β-dihalo γ-methoxycarbonyloxy and γ-acetoxy butenolides. Org. Lett. 2003, 5, 5015–5017. [Google Scholar] [CrossRef]
- Kurbangalieva, A. R.; Devyatova, N. F.; Bogdanov, A. V.; Berdnikov, E. A.; Mannafov, T. G.; Krivolapov, D. B.; Litvinov, I. A.; Chmutova, G. A. Phosphorus, sulfur, silico. 2007, 182, 607–630. [CrossRef]
- Zhang, J.; Sharma, K. D.; Curran, T. T.; Belmont, D. T.; Davidson, J. G. Efficient synthesis of novel γ-substituted γ-butenolides by Lewis acid catalyzed addition of metal enolates of active methylene compounds to mucohalic acids. J. Org. Chem. 2005, 70, 5890–5895. [Google Scholar]
- Zhang, J.; Blazecka, P. G.; Berven, H.; Belmont, D. Metal-mediated allylation of mucohalic acids: facile formation of γ-allylic α,β-unsaturated γ-butyrolactones. Tetrahedron Lett. 2003, 44, 5579–5582. [Google Scholar] [CrossRef]
- Zhang, J.; Blazecka, P. G.; Belmont, D.; Davidson, J. G. Reinvestigation of mucohalic acids, versatile and useful building blocks for highly funtionalized α,β-unsaturated γ-butyrolactones. Org. Lett. 2002, 4, 4559–4561. [Google Scholar] [CrossRef]
- Arayarat, P.; Singh, H.; Lattmann, E. Solid phase synthesis of substituted 4-amino-5-hydroxy-2(5H)-furanones. Sci. Asia 2001, 27, 121–125. [Google Scholar] [CrossRef]
- Jähnisch, K.; Duczek, W. Chemistry of mucohalic acid IV. Reactions of mucochloric acid derivatives with aniline. J. Prakt. Chem. 1990, 332, 117–121. [Google Scholar] [CrossRef]
- Lattmann, E.; Ayuko, W. O.; Kinchinaton, D.; Langley, Ch. A.; Singh, H.; Karimi, L.; Tisdale, M. Synthesis and evaluation of 5-arylated 2(5H)-furanones and 2-arylated pyridazin-3(2H)-ones as anti-cancer agents. J. Pharm. Pharmacol. 2003, 55, 1259–1265. [Google Scholar]
- Estevez, I.; Raviña, E.; Sotelo, E.; Pyridazines, X.V. Synthesis of 6-aryl-5-amino-3(2H)-pyridazinones as potential platelet aggregation inhibitors. J. Heterocycl. Chem. 1998, 6, 1421–1428. [Google Scholar]
- Moore, H. W.; Arnold, M. J. Photolysis of 4-diazopyrrolidine-2,3-diones. A new synthetic route to mono- and bicyclic β-lactams. J. Org. Chem. 1983, 48, 3365–3367. [Google Scholar] [CrossRef]
- Lattmann, E.; Dunn, S.; Niamsanit, S.; Sattayasai, N. Synthesis and antibacterial activities of 5-hydroxy-4-amino-2(5H)-furanones. Bioorg. Med. Chem. Lett. 2005, 15, 919–921. [Google Scholar] [CrossRef]
- Asplund, D.; Kronberg, L.; Sjoholm, R.; Munter, T. Reaction of mucochloric acid with adenosine: formation of 8-(N6-adenosinyl)ethenoadenosine derivatives. Chem. Res. Toxicol. 1995, 8, 841–846. [Google Scholar] [CrossRef]
- Kronberg, L.; Asplund, D.; Maki, J.; Sjoholm, R. Reaction of mucochloric and mucobromic acids with adenosine and cytidine: formation of chloro- and bromopropenal derivatives. Chem. Res. Toxicol. 1996, 9, 1257–1263. [Google Scholar] [CrossRef]
- Zhang, J.; Blazecka, D.; Davidson, J. G. First direct reductive amination of mucochloric acid: a simple and efficient method for preparing highly functionalized α,β-unsaturated γ-butyrolactams. Org. Lett. 2003, 5, 553–556. [Google Scholar] [CrossRef]
- Sarma, K. D.; Zhang, J.; Huang, Y.; Davidson, J. G. Amino acid esters and amides for reductive amination of mucochloric acid: synthesis of novel γ-lactams, short peptides and antiseizure agent Levetiracetam (Keppra*). Eur. J. Org. Chem. 2006, 3730–3737. [Google Scholar]
- Miao, S.; Andersen, R. J. A. Rubrolides A-H, metabolites of the colonial tunicate Ritterella rubra. J. Org. Chem. 1991, 56, 6275–6280. [Google Scholar] [CrossRef]
- Ortega, J. J.; Zubia, E.; Ocaña, J. M.; Naranjo, S.; Salva, J. New rubrolides from the ascidian synoicum blochmanni. Tetrahedron 2000, 56, 3963–3967. [Google Scholar] [CrossRef]
- Surivet, J. P.; Vatale, J. M. Concise total synthesis of (+)-goniofufurone and goniobutenolides A and B. Tetrahedron Lett. 1996, 37, 4373–4376. [Google Scholar] [CrossRef]
- Gondela, E.; Walczak, K. Z. Synthesis and preliminary bioactivity assays of 3,4-dichloro-5-(ω-hydroxyalkylamino)-2(5H)-furanones. Eur. J. Med. Chem. 2010, 45, 3993–3997. [Google Scholar] [CrossRef]
- Jain, K. S.; Chitre, T. S.; Miniyar, P. B.; Kathiravan, M. K.; Bendre, V. S.; Veer, V. S.; Shahane, S. R.; Shishoo, C. J. Biological and medicinal significance of pyrimidines. Curr. Sci. 2006, 90, 793–803. [Google Scholar]
- Dolman, N. P.; More, J. C. A.; Alt, A.; Knauss, J. L.; Troop, H. M.; Bleakman, D.; Collingridge, G. L.; Jane, D. E. Structure-Activity Relationship Studies on N3-Substituted Willardiine Derivatives Acting as AMPA or Kainate Receptor Antagonists. J. Med. Chem. 2006, 49, 2579–2592. [Google Scholar] [CrossRef]
- Bolitt, V.; Mioskowski, Ch. Direct Preparation of 2-Deoxy-D-glucopyranosides from Glucals without Ferrier Rearrangement. J. Org. Chem. 1990, 55, 5812–5813. [Google Scholar] [CrossRef]
- Pierwocha, A.; Walczak, K. The use of tri-O-acetyl-D-glucal and -D-galactal in the synthesis of ω-aminoalkyl 2-deoxy- and 2,3-dideoxy-D-hexopyranosides. Carbohydrate Res. 2008, 343, 2680–2686. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 4a-c are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gondela, E.; Walczak, K.Z. Convenient Synthesis of 3,4-Dichloro-5-hydroxy-2(5H)-Furanone Glycoconjugates. Molecules 2011, 16, 1011-1020. https://doi.org/10.3390/molecules16021011
Gondela E, Walczak KZ. Convenient Synthesis of 3,4-Dichloro-5-hydroxy-2(5H)-Furanone Glycoconjugates. Molecules. 2011; 16(2):1011-1020. https://doi.org/10.3390/molecules16021011
Chicago/Turabian StyleGondela, Edyta, and Krzysztof Z. Walczak. 2011. "Convenient Synthesis of 3,4-Dichloro-5-hydroxy-2(5H)-Furanone Glycoconjugates" Molecules 16, no. 2: 1011-1020. https://doi.org/10.3390/molecules16021011
APA StyleGondela, E., & Walczak, K. Z. (2011). Convenient Synthesis of 3,4-Dichloro-5-hydroxy-2(5H)-Furanone Glycoconjugates. Molecules, 16(2), 1011-1020. https://doi.org/10.3390/molecules16021011