2. Results and Discussion
Liquid-liquid partitioning of the MeOH extract of the seed of L. domesticum cv Kokossan into n-hexane, EtOAc and aqueous MeOH fractions gave the n-hexane fraction (4 g) as the most active one, with 100% antifeedant activity at 1% concentration. Purification of the n-hexane fraction using silica gel 60 open column chromatography led to the isolation of compounds 1 and 2. In addition, the MeOH extract of the bark of L. domesticum was partitioned between n-hexane and ethyl acetate to give the ethyl acetate fraction. A crude ethyl acetate fraction was subjected to vacuum column chromatography on silica gel 60 and further purified by silica gel column chromatography to yield compounds 3–5.
Kokosanolide A (
1) was obtained as a white needle-like crystals, m.p. 178–180 °C, from
n-hexane-EtOAc. Its molecular formula was established to be C
27H
32O
9 by LC-ESI-MS data (
m/z 500.8093, [M+H]
+), which combined with the
1H- and
13C-NMR spectral data (
Table 1), thus indicated 12 degrees of unsaturation. The UV spectrum showed an absorption maximum at 282 nm (ε 4,600), indicating the presence of an α-β-unsaturated ketone. The IR spectrum showed bands which were ascribable to hydroxyl (ν
max 3,427 cm
−1), a ketone (ν
max 1,753 cm
−1), unsaturated ketone (ν
max 1,709 cm
−1), isolated double bond (ν
max 1,631 cm
−1) and
gem-dimethyl (ν
max 1,449 and 1,389 cm
−1) functionalities. The
1H-NMR spectrum showed the presence of three singlets (δ 0.98, 1.07 and 1.37) from tertiary methyl groups and one doublet at δ 1.13 (
J = 8 Hz) from a secondary methyl group which in turn was correlated to H-10 (δ 3.33, 1H, q,
J = 8 Hz). A singlet appeared in the downfield region (δ 3.67) that was ascribed to the C-27 methoxy protons. A detailed analysis of
1H-NMR spectrum showed characteristic signals of a tetranortriterpenoid skeleton with a
β-substituted furan at δ 7.46, 7.40 and 6.43 and the presence of an olefinic signal of an
α,β-unsaturated ketone at δ 6.28 (1H, s) [
3,
4]. The
13C-NMR, together with APT spectra, revealed 27 carbon signals, including characteristic signals due to a furan ring [δ 143.5 (d), 141.9 (d), 120.5 (s) and 110.8 (d)], a ketone (δ 208.5), two ester groups (δ 172.4 and 165.4), one oxygenated carbon (δ 108.4) and
α,
β-unsaturated ketone (δ 168.6 and 116.7), thus suggesting that
1 possesed a hexacyclic structure with a furan moiety. The
1H-
1H COSY spectrum of
1 showed proton correlations of H
5/H
6, H
9/H
8/H
22, H
9/H
11/H
12, H
5/H
10/H
19, and H
24/H
25, supporting the presence of tetranortriterpenoid structure with a furan ring [
3,
4,
5]. The connectivity of these partial structures were established from the HMBC spectral data (
Figure 2).
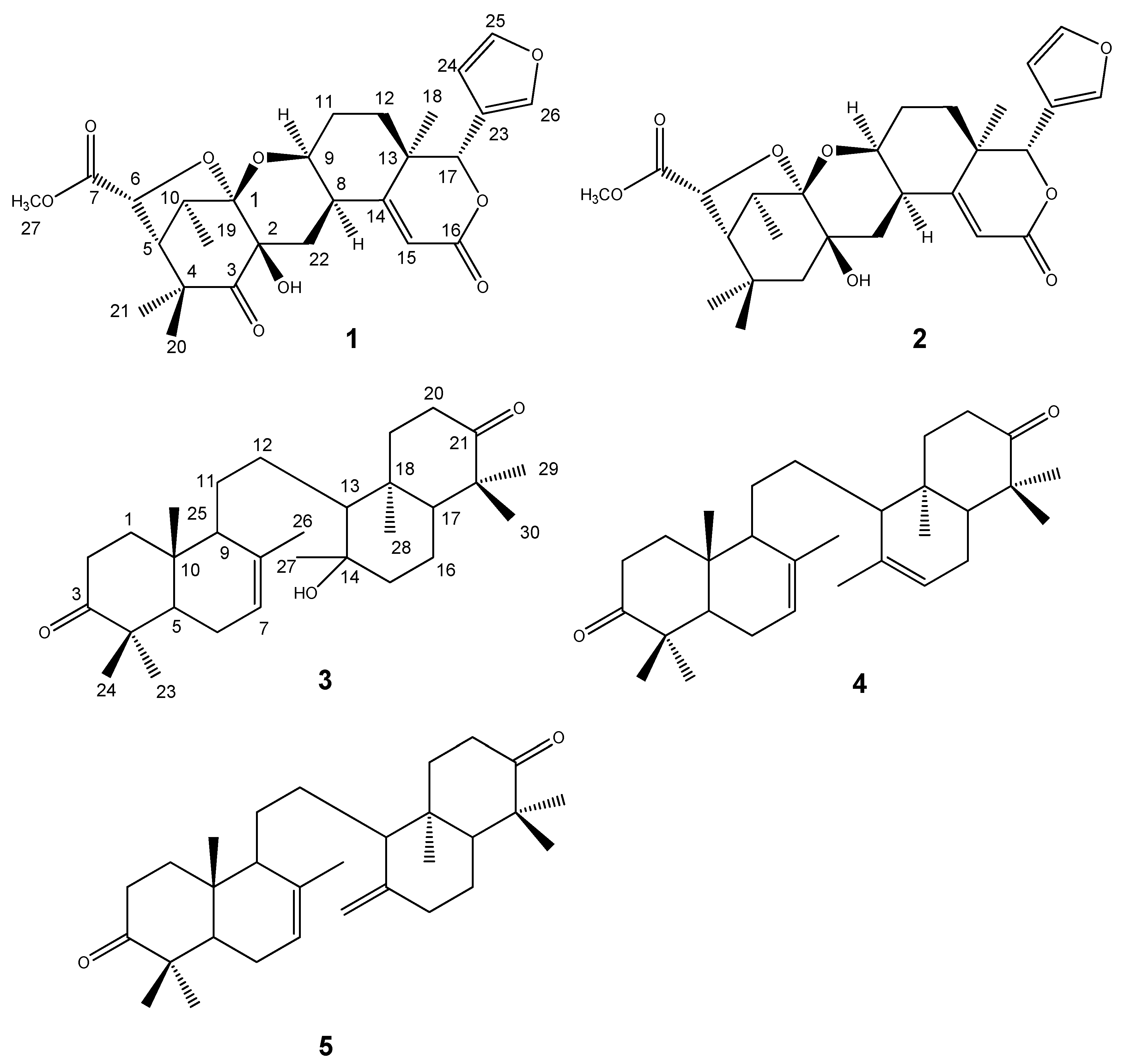
Figure 2.
Selected HMBC (H→C) (a) and NOESY (↔) (b) correlations of kokosanolide A.
Figure 2.
Selected HMBC (H→C) (a) and NOESY (↔) (b) correlations of kokosanolide A.
The oxygenated H-17 (δ 5.17) revealed correlations to C-23 (δ 120.5), C-24 (δ 110.8) and C-26 (δ 141.9), indicating that the furan ring was located at C-17. The signals correlating H-15 (δ 6.28) with C-8 (δ 34.6), C-13(δ 39.6), and C-16 (δ 165.4), suggested the presence of an α,β-unsaturated δ-lactone ring system. The positioning of the pyran ring was established by the correlation signals of H-22 (δH 2.46 and 2.77) with C-1 (δ 108.4), C-2 (δ 76.1), C-9 (δ 69.5), C-8 (δ 34.6) and C-14 (δ 168.6).
Table 1.
NMR spectral data for compounds 1 and 2. a
Table 1.
NMR spectral data for compounds 1 and 2. a
| 1 | 2 |
|---|
| Position | 13C NMR | 1H NMR | HMBC | 13C NMR | 1H NMR |
| δC (mult., ppm) | δH (integral, mult.,
J Hz) | (1H to 13C) | δC (mult., ppm) | δH (integral, mult.,
J Hz) |
| 1 | 108.4 (s) | - | - | 106.9 (s) | - |
| 2 | 76.1 (s) | - | - | 76.1 (s) | - |
| 3 | 208.6 (s) | - | - | 20.9 (t) | 1.71 (1H, d, 6.2) |
| | | | | | 2.18 (1H, m) |
| 4 | 48.4 (s) | - | - | 47.8 (s) | - |
| 5 | 56.4 (d) | 2.27 (1H, dd, 4, 7) | 1, 3, 4, 10 | 55.9 (d) | 2.12 (1H, dd, 3.7, 6) |
| 6 | 77.4 (d) | 4.81 (1H, d, 4) | 4, 5, 7 | 76.7 (d) | 4.82 (1H, d, 4) |
| 7 | 172.4 (s) | - | - | 171.9 (s) | - |
| 8 | 34.6 (d) | 2,64 (1H, dd, 5.3, 6.5) | 14 | 34.4 (d) | 2.31 (1H, m) |
| 9 | 69.5 (d) | 4.39 (1H, m) | 14 | 67.8 (d) | 4.13 (1H, m) |
| 10 | 37.3 (d) | 3.33 (1H, q, 8) | 2, 4, 5, 19 | 36.5 (d) | 3.29 (1H, m) |
| 11 | 25.3 (t) | 1.79 (1H, m) | 8, 12, 13 | 26.8 (t) | 1.76 (1H, m) |
| | | 1.91 (1H, m) | - | | 1.90 (1H, m) |
| 12 | 27.8 (t) | 1.26 (1H, m) | 9, 11, 13, 17 | 29.4 (t) | 1.23 (1H, m) |
| | | 1.74 (1H, m) | - | | 1.72 (1H, m) |
| 13 | 39.6 (s) | - | - | 38.4 (s) | - |
| 14 | 168.6 (s) | - | - | 167.4 (s) | - |
| 15 | 116.7 (d) | 6.28 (1H, s) | 8, 13, 16 | 117.4 (d) | 6.43 (1H, s) |
| 16 | 165.4 (s) | - | - | 165.4 (s) | - |
| 17 | 81.5 (d) | 5.17 (1H, s) | 14, 23, 24, 26 | 81.7 (d) | 5.14 (1H, s) |
| 18 | 18.2 (q) | 1.07 (3H, s) | 12, 13, 14, 17 | 19.6 (q) | 1.26 (3H, s) |
| 19 | 12.2 (q) | 1.13 (3H, d, 8) | 1, 5, 10 | 11.7 (q) | 1.17 (3H, d, 7.8) |
| 20 | 23.7 (q) | 0.98 (3H, s) | 3, 4, 5, 21 | 23.3 (q) | 0.98 (3H, s) |
| 21 | 30.1 (q) | 1.37 (3H, s) | 3, 4, 5, 20 | 30.9 (q) | 1.37 (3H, s) |
| 22 | 25.4 (t) | 2.46 (1H, dd, 7.5, 5.3) | 1, 2, 3, 8, 9, 14 | 25.7 (t) | 2.41 (1H, m) |
| | | 2.77 (1H, dd, 7.5, 4.7) | - | - | 2.70 (1H, m) |
| 23 | 120.5 (s) | - | - | 119.7 (s) | - |
| 24 | 110.8 (d) | 6.43 (1H, d, 4.4) | 23, 25, 26 | 110.2 (d) | 6.45 (1H, s) |
| 25 | 143.5 (d) | 7.40 (1H, d, 4.4) | 23, 24, 26 | 142.8 (d) | 7.40 (1H, s) |
| 26 | 141.9 (d) | 7.47 (1H, s) | 23, 24, 25 | 141.4 (d) | 7.47 (1H, s) |
| 27-OCH3 | 52.6 (q) | 3.67 (3H, s) | - | 52.2 (q) | 3.68 (3H, s) |
Other correlations of methyl signal at δ 0.98 and 1.37 to C-3 (δ 208.6), C-4 (48.4) and C-5 (δ
H 56.4), suggesting that
gem-dimethyl was located at C-4. The carbomethoxyl signal (δ 3.67) and the signal of H-6 (δ 4.81) were correlated to an ester carbonyl (δ 172.4), suggesting that an ester group was located at C-6. The relative configuration of
1 was elucidated by NOESY correlations as shown in
Figure 2. NOESY correlation of H-8/H-9 suggesting that pyran ring should be β−orientation. Correlations between H-6/H-5/H-20 indicated that methyl group and tetrahydrofuran ring should be α-orientation. Thus, the gross structure of tetranortriterpenoid
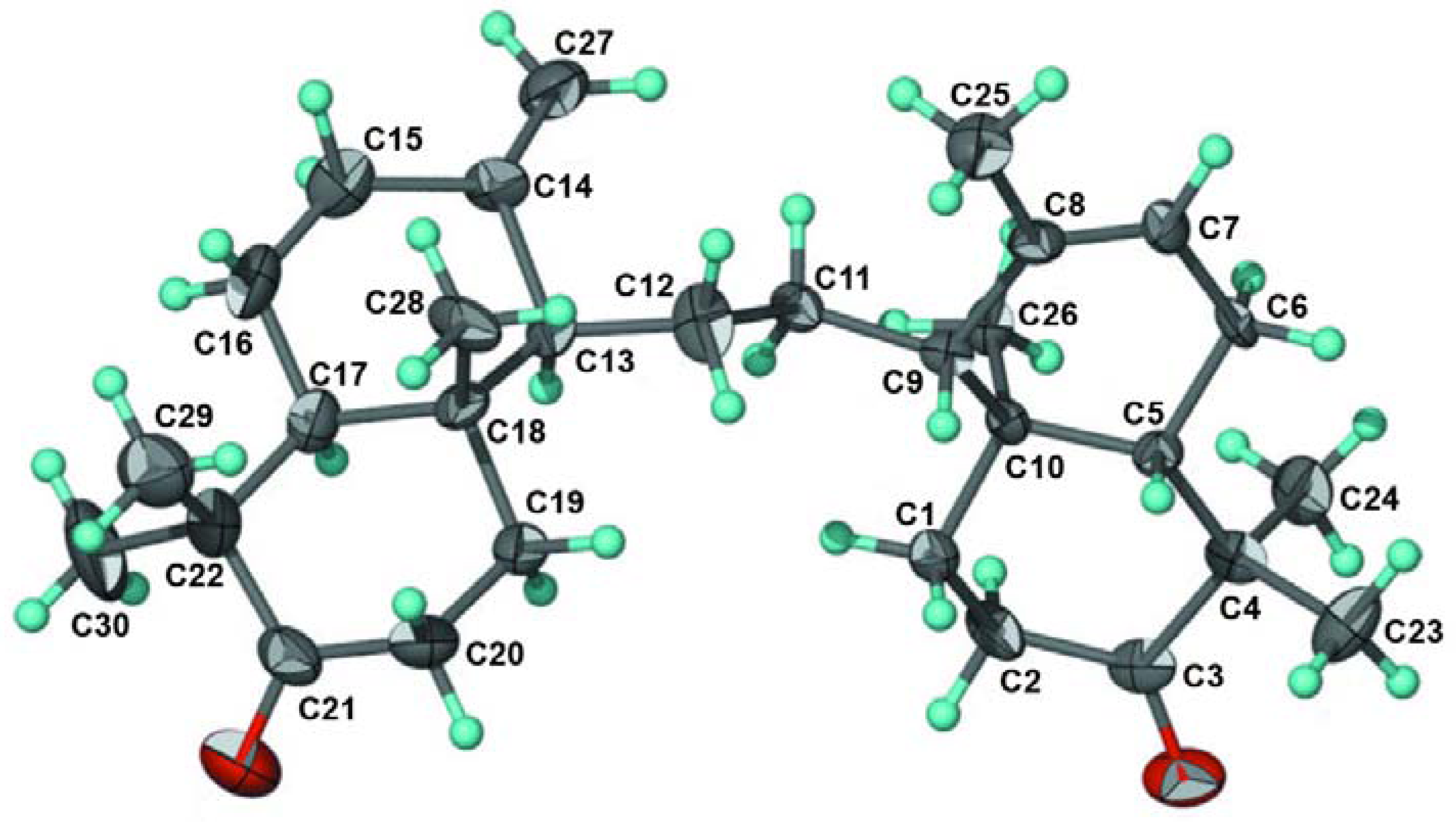
1 was elucidated as a hexacyclic ring system. The structure and relative stereochemistry were further elucidated by using single-crystal X-ray diffraction analysis [
7]. An ORTEP drawing of
1 is shown in
Figure 3. Consequently, the structure of tetranortriterpenoid
1 was established to be a tetranortriterpenoid and was named kokosanolide A.
Figure 3.
ORTEP drawing for kokosanolide A reproduced from Mayanti
et al. [
7].
Figure 3.
ORTEP drawing for kokosanolide A reproduced from Mayanti
et al. [
7].
Kokosanolide C (
2) was obtained as colorless needle-like crystals from
n-hexane-EtOAc and decomposed during the measurement of its melting point. The UV spectrum showed an absorption maximum at 275 nm (ε 4,500), indicating the presence of an α-β-unsaturated ketone. The IR spectrum showed bands which were ascribable to hydroxyl (ν
max 3,563 cm
−1), a ester carbonyl (ν
max 1,758 cm
−1) and unsaturated ketone (ν
max 1,704 cm
−1). The
1H- and
13C-NMR (
Table 1) spectra of
2 were quite similar to those of
1, except for the absence of the ketone signal at δ 208.5 and appearance of a geminal proton signal at [δ
H 2.18 (1H, m), 1.71 (1H, m); δ
C 20.9]. In the HMBC spectrum of
2, long range correlations were observed between the signals at δ 1.72 and 2.18 and the carbon signals at δ 76.1 (C-2), 47.8 (C-4) and 55.9 (C-5), suggesting that compound
2 was a 3-deoxo derivative of compound
1 and it was thus named kokosanolide C.
Kokosanolide B (
3) was obtained as cubic crystals, m.p. 148–150 °C, from
n-hexane-EtOAc. The molecular formula of
3 was determined to be C
30H
48O
3 by LC- ESI-MS data (
m/z 456.6892, [M+H]
+), and combined with the
1H- and
13C-NMR spectral data (
Table 2), thus required seven degrees of unsaturation. IR absorption bands at 3,749, 1,705, 1,384 and 1,261 cm
−1 suggested the presence of hydroxyl, carbonyl, and
gem-dimethyl functionalities, respectively. Analysis of
1H- and
13C-NMR data, DEPT and the HMQC spectra of
3 revealed the presence of thirty signals: three sp
2 and five sp
3 quaternary carbons, one sp
2 and four sp
3 methines, nine sp
3 methylenes, and eight methyl groups. Among them, one sp
2 methine (δ
C 121.7; δ
H (1H, 5.41, m) was ascribed to the isolated double bond, while two carbonyl carbons (δ 216.9 and 217.1 ppm) and one sp
3 carbon (δ 74.0) were assigned to those bearing an oxygen atom (C-3, C-21 and C-14, respectively), suggesting that compound
3 was an onoceranoid-type triterpenoid [
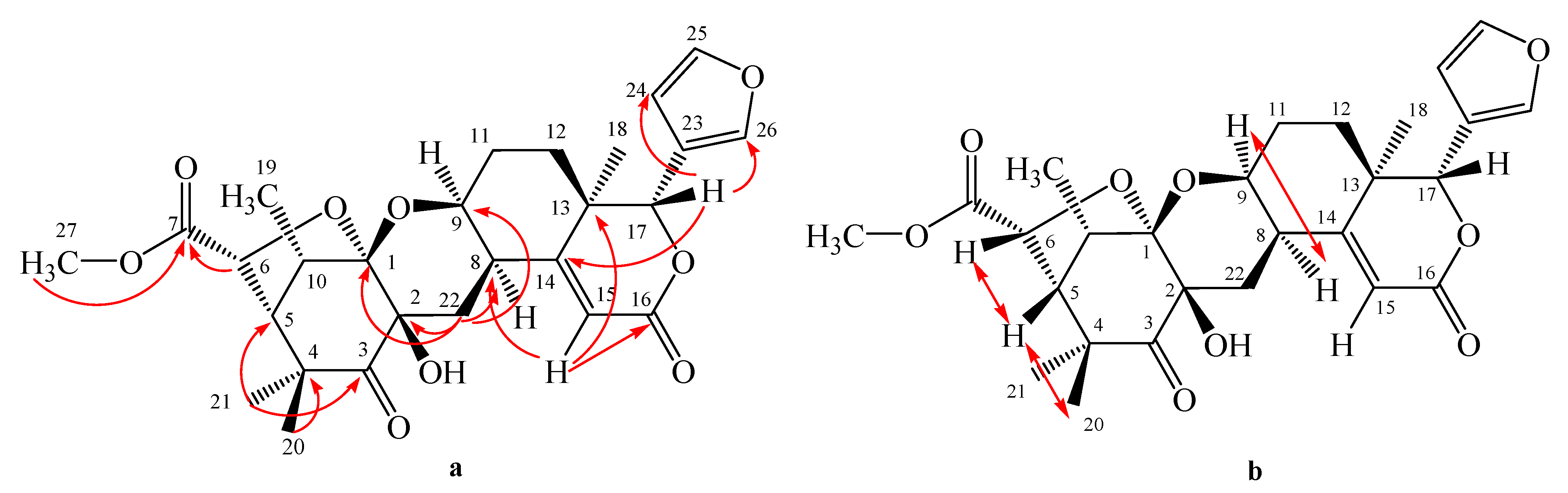
5]. The positions of the ketones, hydroxyl and isolated double bond were further determined by the COSY and HMBC experiments (
Figure 4), H-7 (δ 5.41) showed correlations to C-6 (δ 28.9), C-9 (δ 55.5) and C-8 (δ 135.3), suggesting that an isolated double bond is located at C-7 and C-8. The methylene signals at C-2 (δ 2.23) revealed correlations to carbonyl signal at δ 216.9, whereas H-5 (δ 1.57) was correlated to C-4 (δ 47.6) and C-3 (δ 216.9), indicating that one of the carbonyl moeity and
gem-dimethyl are placed at C-3 and C-4, respectively. Furthermore, the methylene signals at C-20 (δ 2.26) showed correlations to the carbonyl signal at δ 217.1, whereas the methine signal at C-17 (δ 1.42) showed correlations to C-22 (δ 47.6 ) and C-21 (δ 217.1), thus suggesting the other carbonyl group and
gem-dimethyls were located at C-21 and C-22, respectively. An oxygenated tertiary carbon signal was revealed to be C-14 by the correlation between methine signal at C-13 (δ
C 1.12 ) to oxygenated carbon at δ
C 74.0 ppm and correlation between methylene signals at C-15 (δ
C 1.46 ) to oxygenated carbon at δ
C 74.0 ppm. Thus, the structure of onoceranoid-type triterpenoid
3 was determined as 8,14-secogammacera-14-hydroxy-7-ene-3,21-dione and was named kokosanolide B. The structure and relative stereochemistry were also elucidated by using single-crystal X-ray diffraction analysis [
8]. An ORTEP drawing of
3 is shown in
Figure 5.
Figure 4.
Selected HMBC (H→ C) and COSY ( ▬) correlations for kokosanolide B.
Figure 4.
Selected HMBC (H→ C) and COSY ( ▬) correlations for kokosanolide B.
Figure 5.
ORTEP drawing for kokosanolide B reproduced from Supratman
et al. [
8].
Figure 5.
ORTEP drawing for kokosanolide B reproduced from Supratman
et al. [
8].
Table 2.
NMR spectral data for compound 3 and 4. a
Table 2.
NMR spectral data for compound 3 and 4. a
| | 3 | 4 |
|---|
| Position | 13C NMR | 1H NMR | HMBC | 13C NMR | 1H NMR |
| δC (mult., ppm) | δH (integral, mult.,
J Hz) | (1H to 13C) | δC (mult., ppm) | δH (integral, mult.,
J Hz) |
| 1 | 38.5 (t) | 1.91 (1H, m); 2.08 (1H, m) | 2, 3, 10 | 38.5 (t) | 1.46 (1H, m); 2.09 (1H, m) |
| 2 | 34.8 (t) | 2.23 (1H, m); 2.41 (1H, m) | 3 | 34.8 (t) | 2.24 (1H, m); 2.70 (1H, m) |
| 3 | 216.9 (s) | - | - | 216.9 (s) | - |
| 4 | 47.6 (s) | - | - | 47.6 (s) | - |
| 5 | 51.6 (d) | 1.57 (1H, m) | 9, 10, 24 | 51.6 (d) | 1.59 (1H, dd, 5, 7) |
| 6 | 28.9 (t) | 1.12 (1H, m); 2.56 (1H, m) | 7, 10 | 30.1 (t) | 1.33 (1H, dd, 7, 10); 1.24 (1H, dd, 5, 10) |
| 7 | 121.7 (d) | 5.41 (1H, m) | 8 | 122.1 (d) | 5.43 (1H, m) |
| 8 | 135.3 (s) | - | - | 135.3 (s) | - |
| 9 | 55.5 (d) | 1.59 (1H, m) | 26 | 55.6 (d) | 1.65 (1H, m) |
| 10 | 36.6 (s) | - | - | 36.7 (s) | - |
| 11 | 21.5 (t) | 1.61 (1H, m); 2.41 (1H, m) | 9 | 24.2 (t) | 1.93 (1H, m); 2.40 (1H, m) |
| 12 | 21.5 (t) | 1.62 (1H, m); 1.76 (1H, m) | 9 | 24.2 (t) | 1.93 (1H, m); 2.40 (1H, m) |
| 13 | 61.8 (d) | 1.12 (1H, m) | 17 | 55.6 (d) | 1.65 (1H, m) |
| 14 | 74.0 (s) | - | - | 135.3 (s) | - |
| 15 | 44.2 (t) | 1.46 (1H, m); 2.23 (1H, m) | 14, 17 | 122.1 (d) | 5.43 (1H, m) |
| 16 | 31.4 (t) | 1.51 (1H, m); 1.84 (1H, m) | 17 | 30.1 (t) | 1.33 (1H, dd, 7, 10); 1.24 (1H, dd, 5, 10) |
| 17 | 55.2 (d) | 1.42 (1H, m) | 18, 22 | 51.6 (d) | 1.59 (1H, dd, 5, 7) |
| 18 | 36.6 (s) | - | - | 36.7 (s) | - |
| 19 | 38.4 (t) | 1.78 (1H, m); 2.10 (1H, m) | 21 | 38.5 (t) | 1.46 (1H, m); 2.09 (1H, m) |
| 20 | 34.1 (t) | 2.26 (1H, m); 2.73 (1H, m) | 19, 21 | 34.8 (t) | 2.24 (1H, m); 2.70 (1H, m) |
| 21 | 217.1 (s) | - | - | 216.9 (s) | - |
| 22 | 47.6 (s) | - | - | 47.6 (s) | - |
| 23 | 25.1 (q) | 1.04 (3H, s) | 5, 24 | 25.1 (q) | 1.04 (3H, s) |
| 24 | 22.3 (q) | 1.08 (3H, s) | 23 | 22.3 (q) | 1.09 (3H, s) |
| 25 | 13.4 (q) | 0.96 (3H, s) | 5, 9, 10 | 13.5 (q) | 0.97 (3H, s) |
| 26 | 22.3 (q) | 1.77 (3H, s) | 9 | 22.5 (q) | 1.72 (3H, s) |
| 27 | 24.2 (q) | 1.21 (3H, s) | 13, 14, 15 | 22.5 (q) | 1.72 (3H, s) |
| 28 | 15.1 (q) | 0.93 (3H, s) | 13, 17 | 13.5 (q) | 0.97 (3H, s) |
| 29 | 21.4 (q) | 1.02 (3H, s) | 17, 22, 30 | 25.1 (q) | 1.04 (3H, s) |
| 30 | 26.5 (q) | 1.09 (3H, s) | 17, 29 | 22.3 (q) | 1.09 (3H, s) |
8,14-Secogammacera-7,14-diene-3,2-dione (
4) was obtained as a white needle-like crystals, m.p. 143–144 °C, from
n-hexane-EtOAc. The molecular formula of
4 was determined to be C
30H
46O
2 by LC-ESI-MS data (
m/z 438.3745, [M+H]
+), which together with
1H- and
13C-NMR spectral data (
Table 2), requires eight degrees of unsaturation. Compound
4 showed no absorption maxima in the UV spectrum indicating the absence of a conjugated double bond. The IR spectrum showed bands which were ascribed to a ketone (ν
max 1,708 cm
−1), isolated double bond (ν
max 1,662 cm
−1) and
gem-dimethyl (ν
max 1,430 and 1,360 cm
−1). The
13C-NMR spectrum of
4 showed 15 signals, similar to those of kokosanolide B, suggesting that
4 has a symmetrical structure. The essential differences between the NMR spectra of
4 and kokosanolide B consisted of the absence of a hydroxyl group and appearance of a double bond [δ 5.43 (1H, m), δ
H 122.1 and 135.3] and fifteen carbon signals, suggesting that
4 was a dehydroxy derivative of kokosanolide B. In order to determine the connectivity of the partial structure due to a newly double bond, HMBC experiments were carried out. The signal of olefinic proton H-15 (δ 5.43) was correlated to C-14 (δ 135.3), C-13 (δ 55.6) and C-16 (δ 30.1), indicating that a new double bond was located at C-14 and C-15, suggesting that
4 has two similar unit structure. Consequently, compound
4 was identified as a 8,14-secogammacera-7,14-diene-3,21-dione [
5].
Compound (
5) was identified as 8,14-secogammacera-7,14(27)-diene-3,21-dione. It was isolated together with compound
4 with a ratio of 1.5:0.5. The
1H and
13C-NMR spectra of
5 were similar to those of
4, except for the appearance of methylene protons and an sp
2 carbon at C-27 [δ
H 5.12 (1H,
J = 10.5 Hz), 5.45 (1H,
J = 10.5 Hz), δ
C 122.1], indicating that
5 is an isomer of
4. The structure was elucidated by using a single-crystal X-ray diffraction analysis [
9] and the ORTEP drawing for
5 is shown in
Figure 6 below.
Compounds
1–
5 were evaluated for antifeedant activity against the fourth instar larvae of
Epilachna vigintioctopunctata at a concentration of 1%. The antifeedant activities of compounds
1–
5 are shown in
Table 3. Among those compounds, kokosanolide C (
2), lacking the ketone group, showed less antifeedant potency, whereas 8,14-secogammacera-14-hydroxy-7-ene-3,21-dione (
3) having the hydroxyl group showed the strongest activity, thus, suggested that an oxygenated fuctional group was an important structural component for antifeedant activity.
Figure 6.
ORTEP drawing for
5 reproduced from Tjokronegero
et al. [
9].
Figure 6.
ORTEP drawing for
5 reproduced from Tjokronegero
et al. [
9].
Table 3.
Antifeedant activity of compounds 1–5.
Table 3.
Antifeedant activity of compounds 1–5.
| Compound | Activity (%) |
|---|
| Kokosanolide A (1) | 78 |
| Kokosanolide C (2) | 0 |
| Kokosanolide B (3) | 99 |
| 8,14-Secogammacera-7,14-diene-3,21-dione (4) | 85 |
| 8,14-Secogammacera-7,14(27)-diene-3,21-dione(5) | 56 |
3. Experimental
3.1. General
Melting points were measured on an Electrothermal melting point apparatus and are uncorrected. Optical rotations were recorded on a Perkin-Elmer 341 polarimeter. The UV spectra were obtained on a UV Ultraspec 3000 Pro spectrophotometer. The IR spectra were recorded on a Perkin-Elmer 1760X FT-IR in KBr. The mass spectra were recorded with a Mariner Biospectrometry-Finnigan instrument. 1H- and 13C-NMR spectra were obtained with a JEOL JNM A-500 spectrometer using TMS as internal standard. All ORTEP diagrams were obtained from previous reports. Chromatographic separations were carried out on silica gel 60 (Merck). TLC plates were precoated with silica GF254 (Merck, 0.25 mm) and detection was achieved by spraying with 10% H2SO4 in ethanol, followed by heating.
3.2. Plant material
The bark and seed of L. domesticum cv Kokossan were collected in Cililin District, Bandung, West Java Province, Indonesia in March 2006. The plant was identified by the staff of the Laboratory of Plant Taxonomy, Department of Biology, Padjadjaran University, Indonesia. A voucher specimen (No. 10184) was deposited at the herbarium of the Padjadjaran University.
3.3. Antifeedant bioassay
Compounds
1–
5 at a concentration of 1% were subjected to select antifeedant assays on
Solanum nigrum leaves against 4th instars larvae of
Epilachna vigintioctopunctata. Antifeedant activity (%AF) was determined by using the equation %AF = (1 – treatment consumption/control consumption) × 100 [
11]. The antifeedant activities of
1–
5 at 1% concentration are presented in
Table 3.
3.4. Extraction and isolation
Dried seeds of L. domesticum cv kokossan (2 kg) were extracted exhaustively with methanol 12 L at room temperature for 3 days. The resulting methanol extract (84 g) was partitioned between n-hexane (2.5 L) and 10% aqueous methanol (2.5 L) to give an n-hexane soluble fraction (4 g) after removal of the solvent. The n-hexane extract was subjected to column chromatography on silica gel 60 using a n-hexane and dichloromethane (8:2). The fraction eluted with n-hexane-dichloromethane (6:4) was further separated by column chromatography on silica gel (n-hexane-ethyl acetate 7:3) to give 1 (150 mg) and 2 (26 mg).
The dried bark of L. domesticum cv kokossan (3 kg) was extracted exhaustively with methanol 15 L at room temperature for 3 days. The methanol extract (250 g) was partitioned with n-hexane (3 L) and ethyl acetate (3 L) to give an n-hexane soluble fraction (70 g) and an ethyl acetate soluble fraction (40 g). The ethyl acetate fraction was subjected to vacuum column chromatography on silica gel 60 by using a step gradient of n-hexane/ethyl acetate. The fraction eluted with n-hexane:ethyl acetate (80:20) was further separated by column chromatography on silica gel using n-hexane:ethyl acetate (95:5) to yield an active fraction (1.5 g). The active fraction was further chromatographed on silica gel using n-hexane/acetone (90:10) to give 3 (10 mg), 4 (50 mg) and 5 (24 mg).
Kokosanolide A (1). White needle-like crystals; [α]
20D + 85° (
c 0.5, CHCl
3); UV (λ
max in EtOH) 282 nm (ε 4,600); IR (KBr) ν
max 3,427, 1,753, 1,709, 1,631, 1,449 and 1,389 cm
−1.
1H-NMR (CDCl
3, 500 MHz), see
Table 1;
13C-NMR (CDCl
3, 125 MHz), see
Table 1; LC-ESI-MS data (
m/z 500.8093, [M+H]
+).
Kokosanolide C (
2). Colorless needle-like crystals; [α]
20D + 96° (
c 0.5, CHCl
3); UV (λ
max in EtOH) 275 nm (ε 4,500); IR (KBr) ν
max 3,563 cm
−1, 1,758 cm
−1, 17,04 cm
−1.
1H-NMR (CDCl
3, 500 MHz), see
Table 1;
13C-NMR (CDCl
3, 125 MHz), see
Table 1.
Kokosanolide B (
3). cubic crystals; [α]
20D – 18.5° (
c 01.0, MeOH); IR (KBr) ν
max 3,749, 1,705, 1,384 and 1,261 cm
−1.
1H-NMR (CDCl
3, 500 MHz), see
Table 2;
13C-NMR (CDCl
3, 125 MHz), see
Table 2. LC-ESI-MS data (
m/z 456.6892, [M+H]
+).
8,14-Secogammacera-7,14-diene-3,21-dione (
4). Needle-like crystals; [α]
20D − 8° (
c 0.5, MeOH); IR (KBr) ν
max 1,708, 1,662, 1,430, 1,360 cm
−1.
1H-NMR (CDCl
3, 500 MHz), see
Table 2;
13C-NMR (CDCl
3, 125 MHz), see
Table 2. LC-ESI-MS data (
m/z 438.3745, [M+H]
+).
8,14-Secogammacera-7,14(27)-diene-3,21-dione (5). Needle-like crystals; IR (KBr) νmax 1,667, 1,454 and 1,384 cm−1. 1H-NMR (CDCl3, 500 MHz), δH (ppm), 0.95 (3H, s), 0.97 (3H, s), 1.04 (3H, s), 1.08 (3H, s), 1.09 (3H, s), 1.10 (3H, s), 1.20 (1H, m), 1.22 (3H, s), 1.24 (1H, dd, J = 5, 10 Hz), 1.33 (1H, dd, J = 7, 10 Hz), 1.40 (1H, m), 1.46 (1H, m), 1.50 (1H, m), 1.59 (1H, dd, J = 5, 7 Hz), 1.63 (1H, m), 1.65 (1H, m), 1.72 (3H, s), 1.79 (1H, m), 1.83 (1H, m), 1.93 (1H, m), 2.04 (1H, m), 2.09 (1H, m), 2.15 (1H, m), 2.24 (1H, m), 2.35 (1H, m), 2.40 (1H, m), 2.45 (1H, m), 2.70 (1H, m), 5.12 (1H, J = 10.5 Hz), 5.43 (1H, m), 5.45 (1H, J = 10.5 Hz); 13C-NMR (CDCl3, 125 MHz), δC (ppm); 13.5, 22.3, 22.5, 24.2, 25.1, 30.1, 34.8, 34.9, 36.7, 38.5, 47.6. 51.2, 51.6, 55.6, 122.1, 135.3, 216.9, 217.0.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



