Chemically Induced Photoswitching of Fluorescent Probes—A General Concept for Super-Resolution Microscopy

Abstract
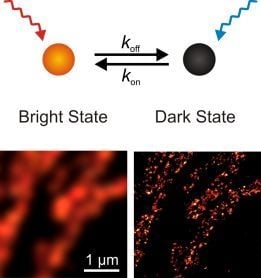
:
1. Introduction
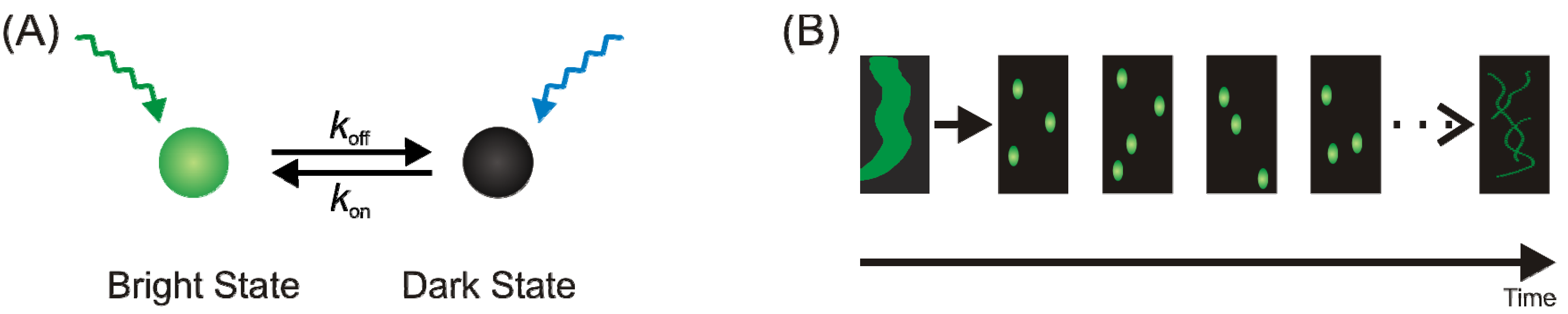


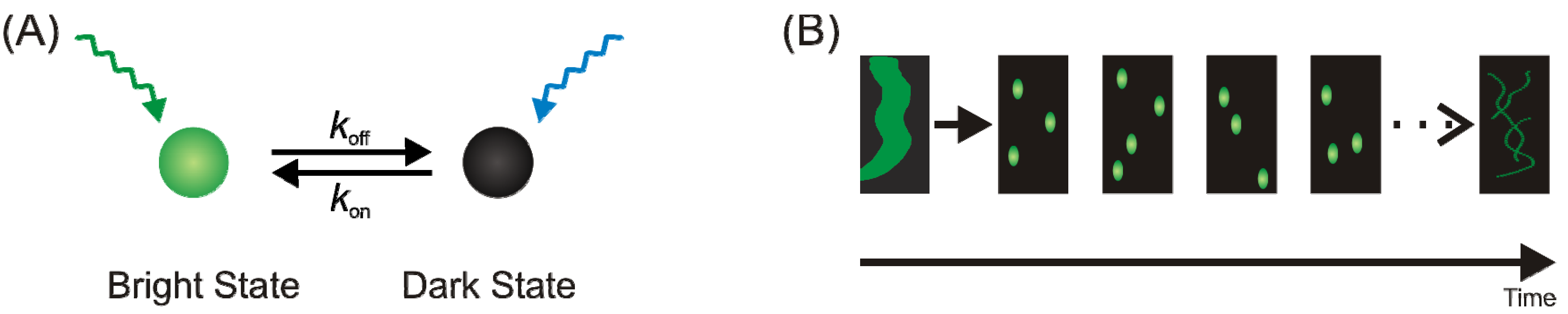
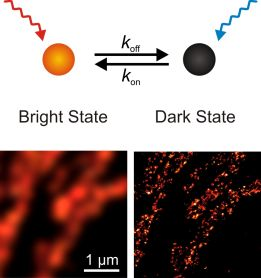{kind=link}
{kind=link}
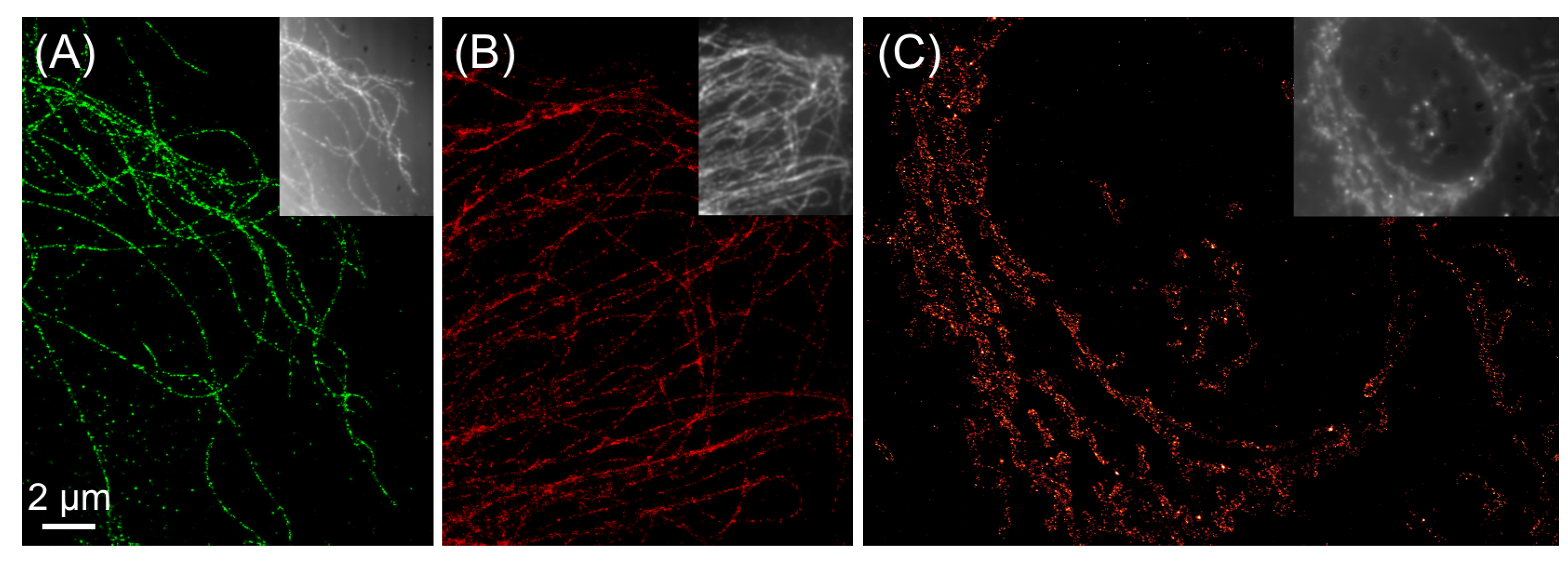
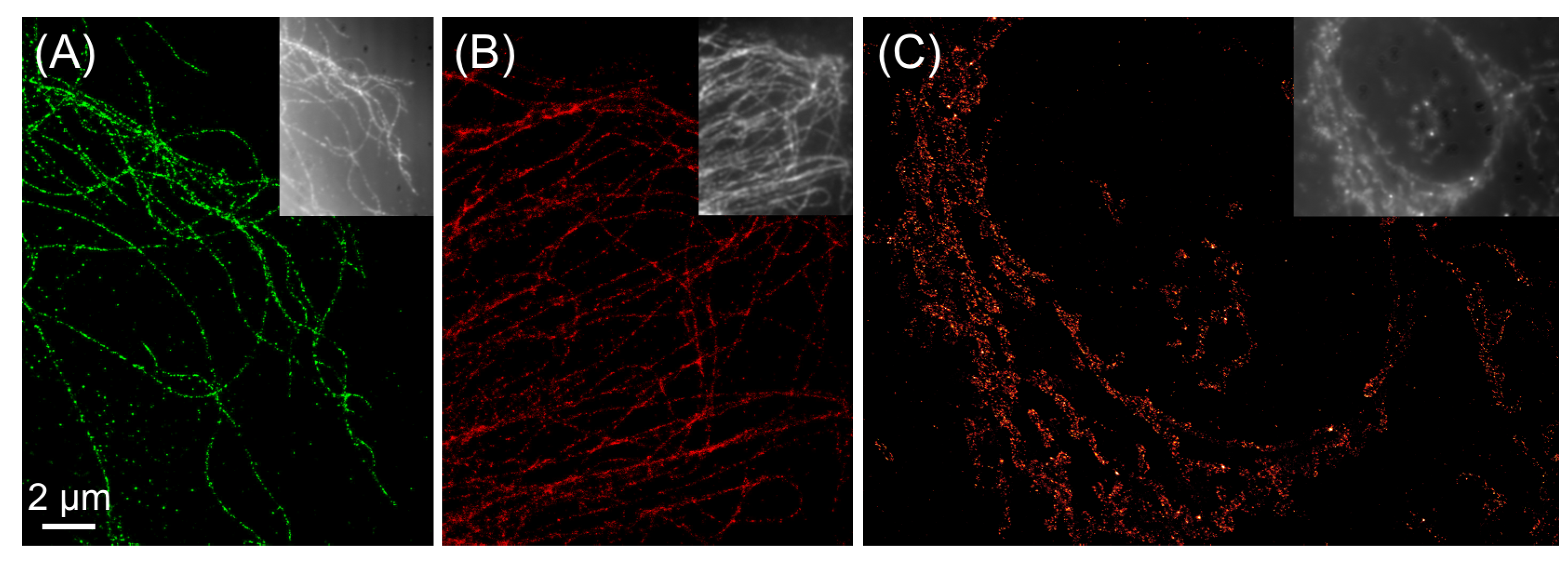{kind=link}
{kind=link}
{kind=link}
{kind=link}
2. Results and Discussion
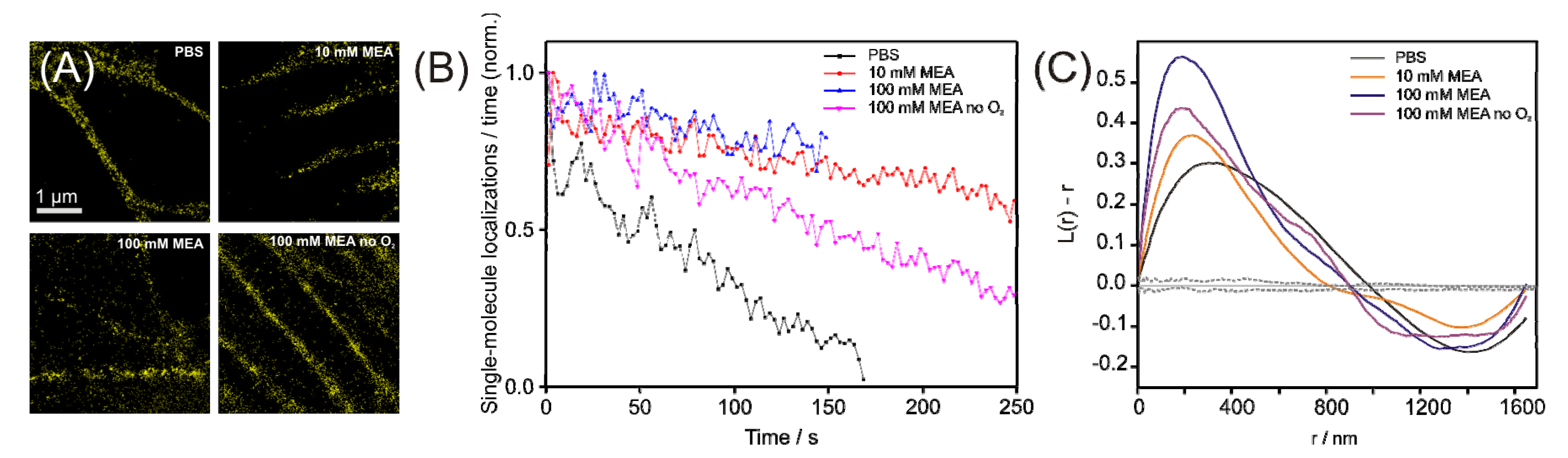
2.1. Operating organic fluorophores as photoswitches using redox chemistry
2.2. Photoswitching of photoactivatable and photoconvertible fluorescent proteins using redox chemistry


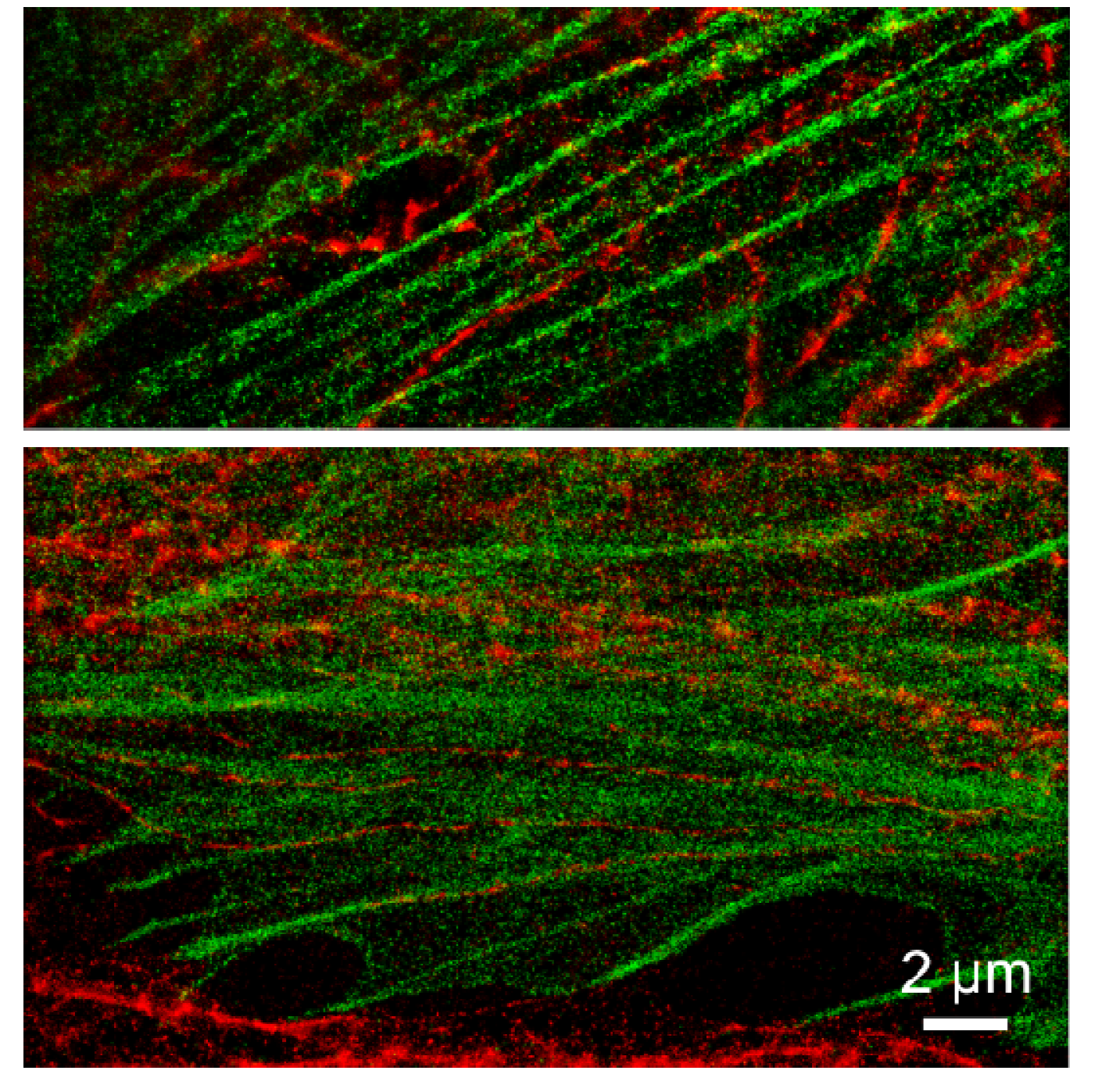
2.3. Prospectives for dual-color localization-based super-resolution imaging


|
|---|
3. Experimental
3.1. Sample preparation
3.2. Microscopic configuration
3.3. PALM imaging
3.4. dSTORM imaging
3.5. Dual color imaging
3.6. Data analysis



4. Conclusions
Supplementary Materials
Acknowledgments
References
- Hell, S.W. Microscopy and its focal switch. Nat. Methods 2009, 6, 24–32. [Google Scholar] [CrossRef]
- Heilemann, M. Fluorescence microscopy beyond the diffraction limit. J. Biotechnol. 2010, 149, 243–251. [Google Scholar]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef]
- Gustafsson, L.; Sparen, P.; Gustafsson, M.; Pettersson, B.; Wilander, E.; Bergstrom, R.; Adami, H.O. Low efficiency of cytologic screening for cancer in situ of the cervix in older women. Int. J. Cancer 1995, 63, 804–809. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar]
- Heilemann, M.; van de Linde, S.; Schuttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008, 47, 6172–6176. [Google Scholar]
- Heilemann, M.; Dedecker, P.; Hofkens, J.; Sauer, M. Photoswitches: Key molecules for subdiffraction-resolution fluorescence imaging and molecular quantification. Laser Photonics Rev. 2009, 3, 180–202. [Google Scholar] [CrossRef]
- Heilemann, M.; Margeat, E.; Kasper, R.; Sauer, M.; Tinnefeld, P. Carbocyanine dyes as efficient reversible single-molecule optical switch. J. Am. Chem. Soc. 2005, 127, 3801–3806. [Google Scholar] [CrossRef]
- Shroff, H.; Galbraith, C.G.; Galbraith, J.A.; Betzig, E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods 2008, 5, 417–423. [Google Scholar]
- van de Linde, S.; Endesfelder, U.; Mukherjee, A.; Schuttpelz, M.; Wiebusch, G.; Wolter, S.; Heilemann, M.; Sauer, M. Multicolor photoswitching microscopy for subdiffraction-resolution fluorescence imaging. Photochem. Photobiol. Sci. 2009, 8, 465–469. [Google Scholar]
- Wombacher, R.; Heidbreder, M.; van de Linde, S.; Sheetz, M.P.; Heilemann, M.; Cornish, V.W.; Sauer, M. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods 2010, 7, 717–719. [Google Scholar]
- Heilemann, M.; van de Linde, S.; Mukherjee, A.; Sauer, M. Super-resolution imaging with small organic fluorophores. Angew. Chem. Int. Ed. Engl. 2009, 48, 6903–6908. [Google Scholar]
- van de Linde, S.; Krstic, I.; Prisner, T.; Doose, S.; Heilemann, M.; Sauer, M. Photoinduced formation of reversible dye radicals and their impact on super-resolution imaging. Photochem. Photobiol. Sci. 2011, 10, 499–506. [Google Scholar]
- Chudakov, D.M.; Verkhusha, V.V.; Staroverov, D.B.; Souslova, E.A.; Lukyanov, S.; Lukyanov, K.A. Photoswitchable cyan fluorescent protein for protein tracking. Nat. Biotechnol. 2004, 22, 1435–1439. [Google Scholar] [CrossRef]
- Klein, T.; Loschberger, A.; Proppert, S.; Wolter, S.; van de Linde, S.; Sauer, M. Live-cell dSTORM with SNAP-tag fusion proteins. Nat. Methods 2011, 8, 7–9. [Google Scholar]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar]
- Gurskaya, N.G.; Verkhusha, V.V.; Shcheglov, A.S.; Staroverov, D.B.; Chepurnykh, T.V.; Fradkov, A.F.; Lukyanov, S.; Lukyanov, K.A. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 2006, 24, 461–465. [Google Scholar] [CrossRef]
- Andresen, M.; Stiel, A.C.; Folling, J.; Wenzel, D.; Schonle, A.; Egner, A.; Eggeling, C.; Hell, S.W.; Jakobs, S. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nat. Biotechnol. 2008, 26, 1035–1040. [Google Scholar]
- Subach, F.V.; Patterson, G.H.; Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J.; Verkhusha, V.V. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods 2009, 6, 153–159. [Google Scholar]
- McKinney, S.A.; Murphy, C.S.; Hazelwood, K.L.; Davidson, M.W.; Looger, L.L. A bright and photostable photoconvertible fluorescent protein. Nat. Methods 2009, 6, 131–133. [Google Scholar]
- Kottke, T.; van de Linde, S.; Sauer, M.; Kakorin, S.; Heilemann, M. Identification of the Product of Photoswitching of an Oxazine Fluorophore Using Fourier Transform Infrared Difference Spectroscopy. J. Phys. Chem. Lett. 2010, 1, 3156–3159. [Google Scholar] [CrossRef]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef]
- Thompson, R.E.; Larson, D.R.; Webb, W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002, 82, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, U.; van de Linde, S.; Wolter, S.; Sauer, M.; Heilemann, M. Subdiffraction-resolution fluorescence microscopy of myosin-actin motility. ChemPhysChem 2010, 11, 836–840. [Google Scholar] [CrossRef]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar]
- Annibale, P.; Scarselli, M.; Kodiyan, A.; Radenovic, A. Photoactivatable Fluorescent Protein mEos2 Displays Repeated Photoactivation after a Long-Lived Dark State in the Red Photoconverted Form. J. Phys. Chem. Lett. 2010, 1, 1506–1510. [Google Scholar] [CrossRef]
- Ripley, B.D. Modelling spatial patterns. J. Roy. Statist. Soc. B 1977, 39, 172–212. [Google Scholar]
- Lillemeier, B.F.; Mortelmaier, M.A.; Forstner, M.B.; Huppa, J.B.; Groves, J.T.; Davis, M.M. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 2010, 11, 90–96. [Google Scholar]
- Wolter, S.; Schuttpelz, M.; Tscherepanow, M.; van de Linde, S.; Heilemann, M.; Sauer, M. Real-time computation of subdiffraction-resolution fluorescence images. J. Microsc. 2010, 237, 12–22. [Google Scholar] [CrossRef]
- Churchman, L.S.; Okten, Z.; Rock, R.S.; Dawson, J.F.; Spudich, J.A. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc. Natl. Acad. Sci. USA 2005, 102, 1419–1423. [Google Scholar]
- Haase, P. Spatial Pattern-Analysis in Ecology Based on Ripley K-Function - Introduction and Methods of Edge Correction. J. Vegetation Sci. 1995, 6, 575–582. [Google Scholar] [CrossRef]
- Ripley, B.D. Tests of `randomness' for spatial point patterns. J. Roy. Statist. Soc. B 1979, 41, 368–374. [Google Scholar]
- Samples Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Endesfelder, U.; Malkusch, S.; Flottmann, B.; Mondry, J.; Liguzinski, P.; Verveer, P.J.; Heilemann, M. Chemically Induced Photoswitching of Fluorescent Probes—A General Concept for Super-Resolution Microscopy. Molecules 2011, 16, 3106-3118. https://doi.org/10.3390/molecules16043106
Endesfelder U, Malkusch S, Flottmann B, Mondry J, Liguzinski P, Verveer PJ, Heilemann M. Chemically Induced Photoswitching of Fluorescent Probes—A General Concept for Super-Resolution Microscopy. Molecules. 2011; 16(4):3106-3118. https://doi.org/10.3390/molecules16043106
Chicago/Turabian StyleEndesfelder, Ulrike, Sebastian Malkusch, Benjamin Flottmann, Justine Mondry, Piotr Liguzinski, Peter J. Verveer, and Mike Heilemann. 2011. "Chemically Induced Photoswitching of Fluorescent Probes—A General Concept for Super-Resolution Microscopy" Molecules 16, no. 4: 3106-3118. https://doi.org/10.3390/molecules16043106
APA StyleEndesfelder, U., Malkusch, S., Flottmann, B., Mondry, J., Liguzinski, P., Verveer, P. J., & Heilemann, M. (2011). Chemically Induced Photoswitching of Fluorescent Probes—A General Concept for Super-Resolution Microscopy. Molecules, 16(4), 3106-3118. https://doi.org/10.3390/molecules16043106