2. Results and Discussion
Heating 3-amino-3-thioxopropanamide (monothiomalonamide,
1) [
10] with ethyl acetoacetate in a refluxing ethanolic potassium hydroxide solution led to the formation of 6-hydroxy-4-methyl-2-thioxo-2,3-dihydropyridine-3-carboxamide (
2) (
Scheme 1).
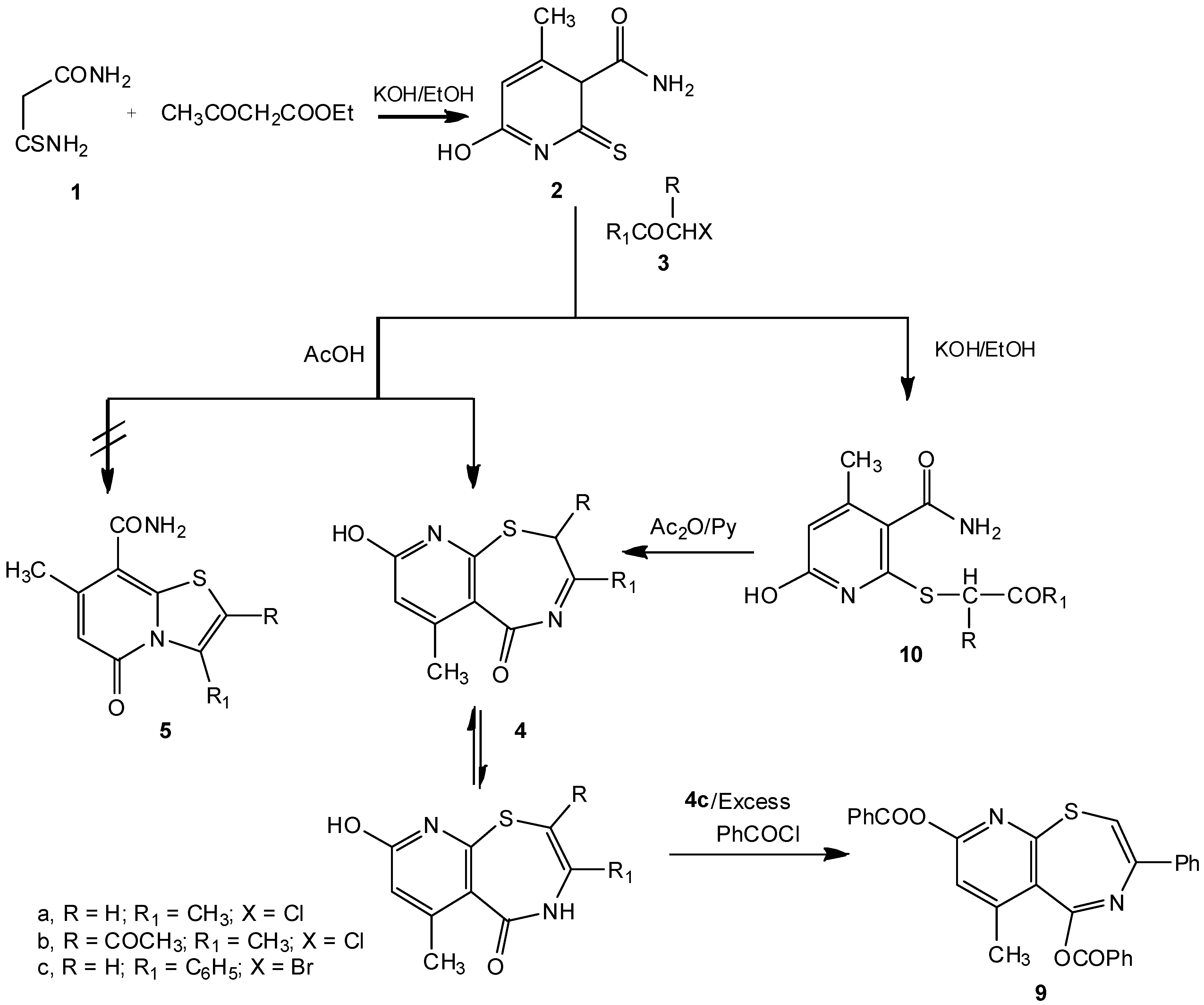
Compound
2 reacted with α-haloketones
3, namely chloroacetone (
3a), 3-chloropentane-2,4-dione (
3b) and phenacyl bromide (
3c), in boiling acetic acid to give directly 2,3-disubstituted-8-hydroxy-6-methyl-2
H,5
H-pyrido[3,2-f][1,4]thiazepin-5-ones
4a-c rather than the isomeric thiazolopyridine derivatives
5a-c (
Scheme 1). The preference for structures
4 over
5 was shown by the IR,
1H-NMR and
13C-NMR spectra. Thus, the IR spectra of compounds
4a,c displayed only one carbonyl absorption near 1,660 cm
−1, whereas structure
5 would display two carbonyl absorptions. The
1H-NMR and
13C-NMR (DMSO-d
6) of
4c, as an example, showed a singlet signal at δ 3.66 ppm and a signal at δ 35.3 ppm, respectively, that correspond to z CH
2 group that would not appear for structure
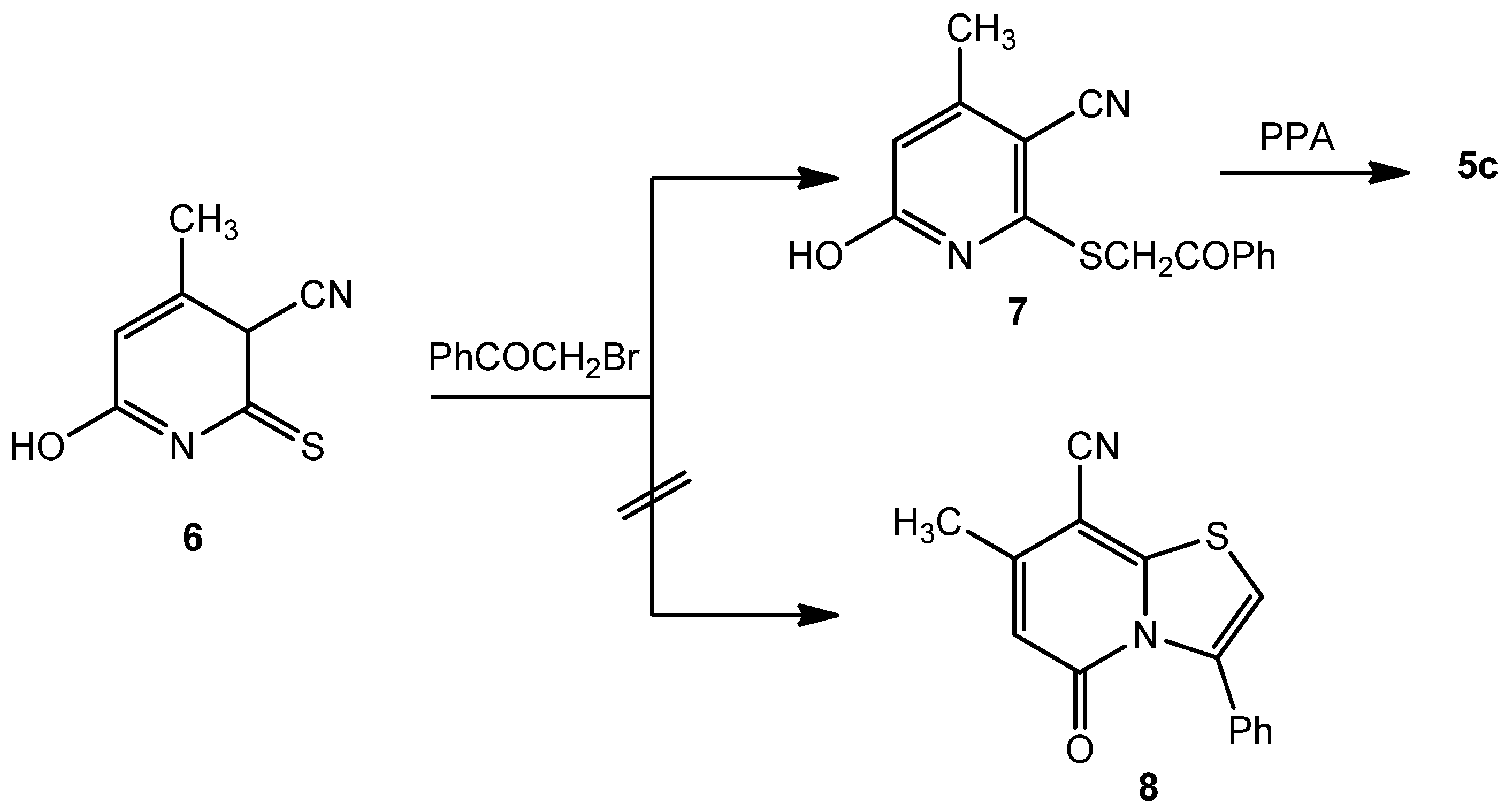
5c. Structure
5c was independently obtained by heating under reflux a mixture of 6-hydroxy-4-methyl-2-thioxo-2,3-dihydropyridine-3-carbonitrile (
6) [
11] with
3c in aqueous ethanolic potassium hydroxide solution to afford the phenacylthiopyridine-3-carbonitrile derivative
7, which was converted to
5c by heating with polyphosphoric acid on a boiling water bath.
This product was found to be different in all aspects (m.p., mixed m.p. and IR) from the reaction product
4c, obtained by the reaction between
2 and
3c in acetic acid. Our original aim was to synthesize the thiazolopyridine-8-carbonitrile
8 by reacting
6 with phenacyl bromide in polyphosphoric acid, and then to hydrolyze product
8 to obtain
5c. Instead we found that hydrolysis of the carbonitrile group into carboxamide took place directly and
5c was eventually obtained (
Scheme 2). A further confirmation of structure
4 could be obtained by reacting
4c, as a representative, with an excess of benzoyl chloride in pyridine to obtain the dibenzoate derivative
9. It is clear that benzoylation is only possible for compound
4c and not for structure
5. When
2 was reacted with each of
3a-c by boiling in ethanolic potassium hydroxide, the S-alkylated derivatives
10a-c were obtained in poor yields. Compounds
10a-c could be cyclized by heating with acetic anhydride in pyridine to give
4a-c, respectively (
Scheme 1).
Our research group has recently [
12] been interested in performing synthesis of some heterocyclic compounds under the environmentally friendly, time saving microwave-assisted conditions. Accordingly, we re-synthesized the previously described compounds
2,
4a-c,
9 and
10a-c under microwave conditions, aiming to increase reaction yields and reduce the reaction times. The results of these preparation indicated that reaction yields increased by 20–30% compared to the traditional conditions. Reaction times were also significantly reduced.
Table 1 summarizes the benefits of using microwave conditions for the synthesis of the above-mentioned compounds.
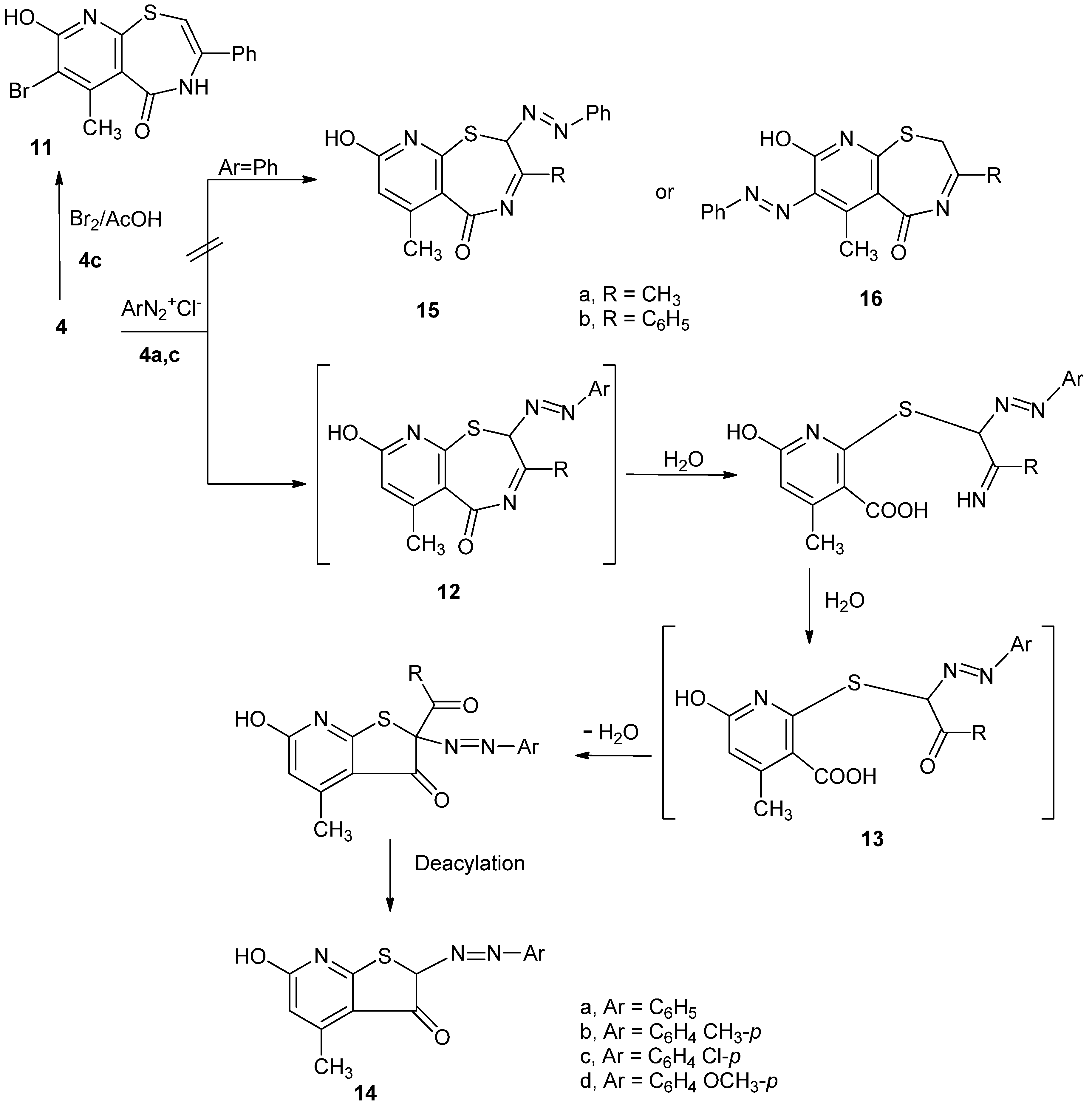
Compound
4c underwent bromination upon treatment with bromine in acetic acid to give 7-bromo-6-methyl-3-phenyl-4,5,8,9-tetrahydropyrido[3,2-f][1,4]thiazepine-5,8-dione (
11) (
Scheme 3) The occurrence of bromination at C7 was proved by studying
1H-NMR spectrum of
11, which revealed the disappearance of the signal that originally existed in
4c at δ6.30 ppm. On the other hand, coupling of
4a-c with arenediazonium chlorides in pyridine/potassium hydroxide took place in an unexpected manner to produce
14a-c.
As an example, treatment of
4a or
4c with benzenediazonium chloride gave one and the same reaction product. The mass spectrum and elemental analysis of the reaction product showed that its molecular formula was C
14H
11N
3O
2S, and could be formulated to be 6-hydroxy-4-methyl-2-(phenylazo)thieno[2,3-b]pyridin-3(2
H)-one (
14a). If coupling took place at either C2 or C7 with no further modification, we would have obtained compounds
15a or
16a, respectively, each of which has the molecular formula C
16H
14N
4O
2S or
15b or
16b, which would have the molecular formula C
21H
16N
4O
2S. As a possible sequence for the formation of
14a from
4a,c, the latter compounds firstly underwent coupling at position 2 to give the corresponding 2-arylazo derivative,
12. Compound
12 underwent hydrolysis and gave
13 which underwent cyclization followed by deacylation to give
14a, as shown in
Scheme 3.
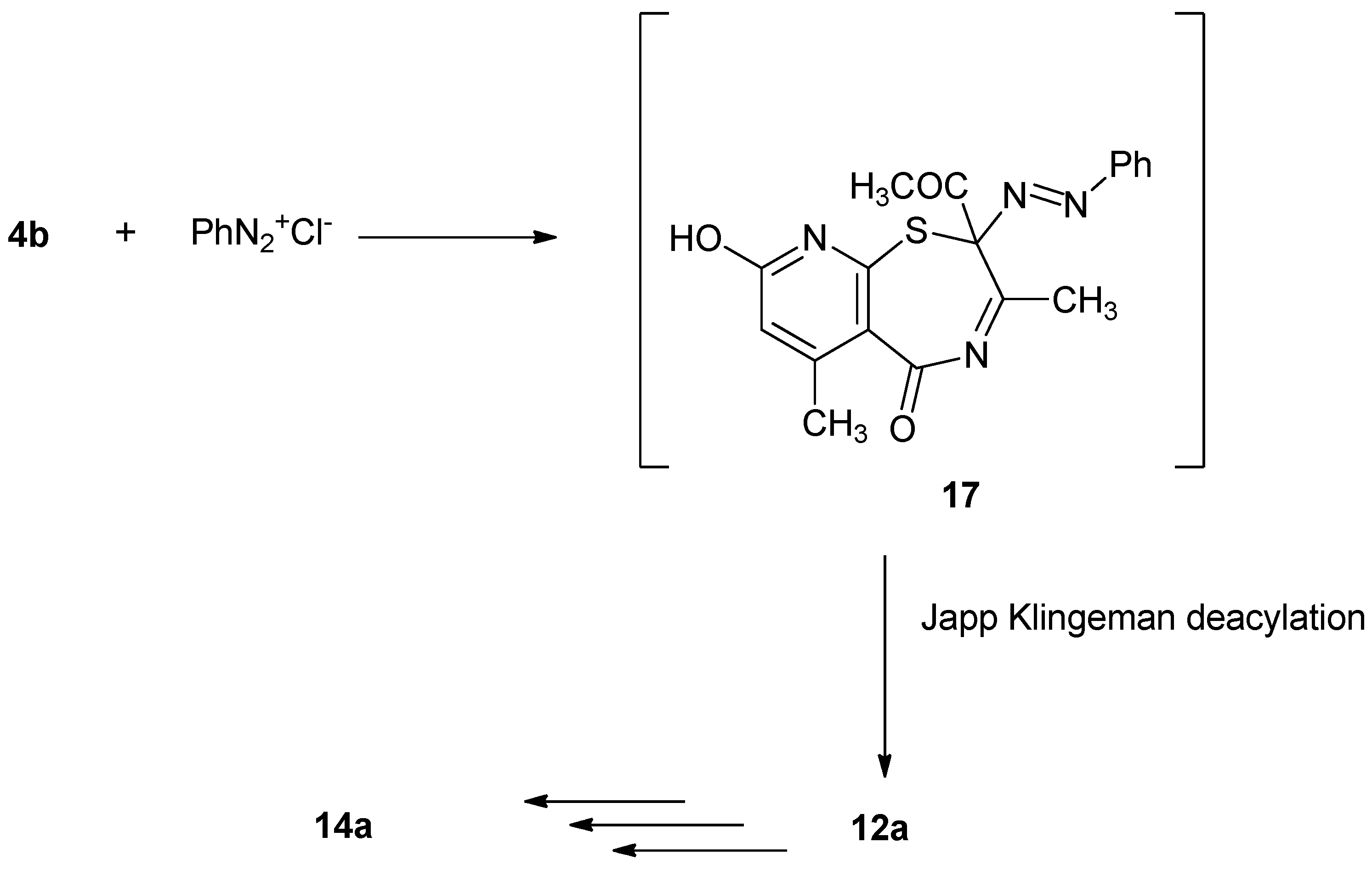
Surprisingly, coupling of
4b with benzenediazonium chloride yielded the same reaction product
14a. The reaction may have proceeded firstly via coupling at position 2 to give the corresponding 2-phenylazo derivative
17 which underwent deacylation (Japp-Klingemann reaction) to give compound
12a. The latter compound underwent hydrolysis then cyclization followed by deacylation to give
14a as described in
Scheme 4.
3. Experimental
3.1. General
Melting points were determined in open glass capillaries on a Gallenkamp melting point apparatus and are uncorrected. IR spectra (KBr discs) were recorded on a Shimadzu FTIR-8201PC spectrophotometer.
1H-NMR and
13C-NMR spectra were recorded on a Varian Mercury 300 MHz and Varian Gemini 200 MHz spectrometers using TMS as an internal standard and DMSO-d
6 as solvent. Chemical shifts were expressed as δ (ppm) units. Mass spectra were recorded at 70 eV on a Shimadzu GCMS-QP1000EX using an inlet type injector. All reactions were followed by TLC (silica gel, aluminum sheets 60 F254, Merck). The Microanalytical Center of Cairo University performed the microanalyses. Microwave reactions were performed with a Millstone Organic Synthesis Unit (MicroSYNTH with touch control terminal) with a continuous focused microwave power delivery system in a pressure glass vessel (10 mL) sealed with a septum under magnetic stirring. The temperature of the reaction mixture was monitored using a calibrated infrared temperature control under the reaction vessel, and control of the pressure was performed with a pressure sensor connected to the septum of the vessel. 3-Amino-3-thioxopropanamide (monothiomalonamide,
1) was prepared according to a literature procedure [
10].
3.2. 6-Hydroxy-4-methyl-2-thioxo-2,3dihydropyridine-3-carboxamide (2)
Method A: A mixture of 3-amino-3-thioxopropanamide (1, 1.18 g, 0.01 mol) and ethyl acetoacetate (1.30 g, 0.01 mol) was heated under reflux in ethanolic potassium hydroxide solution (0.56 g, 0.01 mole in 20 mL ethanol) for 3 h. The formed white potassium salt was dissolved in water and acidified with hydrochloric acid. The precipitated solid was filtered off, washed with ethanol, dried and recrystallized from ethanol.
Method B: The same reactants of method A were heated in the microwave apparatus at 500 w and 140 °C for 15 min. The reaction mixture was treated in a similar manner to method A to afford compound 2. Compound 2 was obtained as white crystals in 55% yield (Method A) and 82% yield (Method B), m.p. 194–195 °C. 1H-NMR: 2.30 (s, 3H, CH3), 3.85 (br s, 1H, OH, D2O exchangeable), 5.24 (s, 1H, H-3), 6.10 (s, 1H, H-5), 7.40 (br s, 2H, NH2, D2O exchangeable); 13C-NMR: 20.3 (CH3), 71.5 (C-3), 112.1, 137.3, 162.3 (sp2 C), 173.1 (CO), 188.0 (CS); IRν: 3307, 3175 (NH, OH) and 1648 (CO); MS: M+ m/z 184.0 (21.1%); Anal. Calcd. for C7H8N2O2S (184.22): C (45.64%), H (4.38%), N (15.21%), S (17.41%). Found: C (45.70%), H (4.20%), N (15.10%), S (17.30%).
3.3. 2,3-disubstituted-8-hydroxy-6-methyl-2H,5H-pyrido[3,2-f][1,4]thiazepin-5-ones 4a-c
Method A: A mixture of 2 (1.84 g, 0.01 mole) and α-haloketones 3a-c (0.01 mole) was heated under reflux in glacial acetic acid (30 mL) for 10 hours. The reaction mixture was then poured onto water; the deposited precipitate was filtered off, dried and crystallized from the proper solvent.
Method B: The same reactants of method A were heated in microwave at 500 w and 140 °C for 30 min. The reaction mixture was treated in a similar manner to method A to give compounds 4a-c.
3,6-Dimethyl-8-hydroxy-2H,5H-pyrido[3,2-f][1,4]thiazepin-5-one (4a). Crystallized from ethanol as white crystals in 61% yield (Method A) and 85% yield (Method B); m.p. 286–287 °C. 1H-NMR: 2.36 (s, 3H, CH3), 2.49 (s, 3H, CH3), 3.66 (s, 2H, CH2), 6.23 (s, 1H, pyridine-H), 12.42 (s, 1H, OH, D2O exchangeable). 13C-NMR: 20.1 (CH3), 21.8 (CH3), 34.7 (CH2), 114.1, 116.9, 145.2, 155.1, 161.2, 165.3 (sp2 C), 179.8 (CO). IRν: 3430 (OH) and 1655 (CO). MS: M+ m/z: 222 (5.7%); Anal. Calcd for C10H10N2O2S (222.26): C (54.05%), H (4.53%), N (12.60%), S (14.43%). Found: C (54.00%), H (4.30%), N (12.50%), S (14.60%).
(3,6-Dimethyl-4,5-dihydro-8-hydroxy-5-oxopyrido[3,2-f][1,4]thiazepin-2-yl)ethanone (4b). Crystallized from acetic acid as white crystals in 59% yield (Method A) and 80% yield (Method B); m.p. 270–271 °C. 1H-NMR: 2.36 (s, 3H, CH3), 2.46 (s, 3H, CH3), 2.54 ppm (s, 3H, CH3), 6.22 (s, 1H, pyridine-H), 12.45 (br.s, 2H, OH, NH, D2O exchangeable); 13C-NMR: 20.3 (CH3), 20.8 (CH3), 28.1 (CH3), 59.0 (C-2), 114.9, 117.0, 145.6, 155.4, 163.2, 165.6 (sp2 C); IRν: 3429 (NH) and 1732 & 1659 (2 CO). MS: M+ m/z: 264 (1.3%); Anal. Calcd for C12H12N2O3S (264.30): C (54.53%), H (4.58%), N (10.60%), S (12.13%). Found: C (54.60%), H (4.30%), N (10.50%), S (12.20%).
8-Hydroxy-6-methyl-3-phenyl-2H,5H-pyrido[3,2-f][1,4]thiazepin-5-one (4c). Crystallized from acetic acid as yellowish white crystals in 56% yield (Method A) and 85% yield (Method B); m.p. 330–331 °C. 1H-NMR: 2.55 (s, 3H, CH3), 3.50 (s, 2H, CH2), 6.30 (s, 1H, pyridine-H), 7.65-7.80 (m, 3H, Ar-H), 7.90–8.00 (m, 2H, Ar-H), 12.65 (br s, 1H, OH, D2O exchangeable). 13C-NMR: 20.1 (CH3), 35.3 (CH2), 114.1, 117.2, 128.3, 129.0, 132.1, 134.5, 145.2, 155.3, 162.7, 165.2 (sp2 C), 180.3 (CO); IRν: 3440 (broad, OH), 2924 (CH) and 1690 (CO). MS: M+ m/z: 284 (100%); Anal. Calcd for C15H12N2O2S (284.33): C (63.36%), H (4.25%), N (9.85%), S (11.28%); Found: C (63.10%), H (4.40%), N (10.00%), S (11.40%).
3.4. 6-Hydroxy-4-methyl-2-((2-oxo-2-phenylethyl)thio)pyridine-3-carbonitrile (7)
Compound
6 (1.66 g, 0.01 mol, [
11]) was dissolved in a warm ethanolic potassium hydroxide solution [prepared by dissolving 0.56 g (0.01 mol) of potassium hydroxide in 50 mL of ethanol]. Then
3c (1.99 g, 0.01 mol) was added. The mixture was refluxed for two hours, then cooled and poured into water. The solid so-formed was filtered off and recrystallized from dilute dimethylformamide to obtain white crystals in 73% yield, m.p. 211–212 °C.
1H-NMR: 2.43 (s, 3H, CH
3), 4.70 (s, 2H, CH
2), 6,11 (s, 1H, pyridine-H), 7.55–7.94 (m, 5H, Ar-H), 12.13 (s, 1H, OH, D
2O exchangeable);
13C-NMR: 21.2 (CH
3), 38.7 (CH
2), 116.1 (CN), 117.0, 117.8, 128.6, 129.3, 133.1, 135.7, 152.2, 156.6, 159.0 (sp2 C), 190.3 (CO); IRν: 2980–3215 (broad, OH), 2180 (CN), 1710 (CO); MS: M
+ m/
z: 284 (15%). Anal. Calcd. for C
15H
12N
2O
2S (284.33): C (63.36%), H (4.25%), N (9.85%), S (11.28%). Found: C (63.10%), H (4.20%), N (9.70%), S (11.40%).
3.5. 7-methyl-5-oxo-3-phenyl-5H-thiazolo[3,2-a]pyridine-8-carboxamide (5c)
Compound 7 (1 g) was heated in polyphosphoric acid (3 g phosphorus pentoxide in 15 mL ortho- phosphoric acid) at 140 °C for three hours. Then the reaction mixture was allowed to cool and poured into water. The solid product so-precipitated was filtered off, washed with water and recrystallized from dimethylformamide to form 5c, obtained as white crystals in 60% yield, m.p. 349–350 °C. 1H-NMR: 2.13 (s, 3H, CH3), 6.15 (s, 1H, pyridine-H), 6.35 (s, 1H, thiazolo-H), 7.30–7.55 (m, 5H, Ar-H), 7.70 (s, 2H, NH2, D2O, exchangeable); 13C-NMR: 21.3 (CH3), 106.0, 118.4, 127.3, 128.5, 128.9, 135.4, 138.1, 145.1, 146.0 (sp2 C), 158.8 (CO), 173.0 (CO); IRν: 3422, 3131 (NH2), 1673, 1650 (2 CO); MS: M+ m/z: 284 (55%). Anal. Calcd. for C15H12N2O2S (284.34): C (63.36%), H (4.25%), N (9.85%), S (11.28%). Found: C (63.50%), H (4.40%), N (10.0%), S (11.40%).
3.6. 6-Methyl-3-phenylpyrido[3,2-f][1,4]thiazepine-5,8-dibenzoate (9)
Method A: To a solution of 4c (2.84 g, 0.01 mol) in pyridine (50 mL), benzoyl chloride (0.02 mol) was added drop-wise with stirring at room temperature. The mixture was then refluxed for three hours and then diluted with cold water (30 mL). The resultant crude product thus precipitated was collected by filtration, washed with water, dried and crystallized from dioxane to afford 9.
Method B: The same reactants of method A were heated in microwave at 500 w and 140 °C for 15 min. The reaction mixture was treated in a similar manner to method A to obtain compound 9. Compound 9 was obtained as white crystals in 57% yield (Method A) and 79% yield (Method B), m.p. 150–151 °C. 1H-NMR: 2.30 (s, 3H, CH3), 6.25 (s, 1H, pyridine-H), 7.30–8.20 (m, 16H, Ar-H); 13C-NMR: 19.8 (CH3), 106.3, 118.3, 119.4, 126.7, 127.1, 128.8, 130.3, 133.8, 134.9, 137.1, 145.0, 155.7, 158.3, 160.0 (sp2 C), 163.4 (CO), 166.0 (CO). IRν: 3144 (CH), 2950 (CH), 1739 (CO). MS: M+ m/z: 492 (2%). Anal. Calcd for C29H20N2O4S (492.55): C (70.72%), H (4.09%), N (5.69%), S (6.51%). Found: C (70.80%), H (4.30%), N (5.80%), S (6.70%)
3.7. 2-[(1,2-Disubstituted)oxoethylthio]-6-hydroxy-4-methylpyridine-3-carboxamides 10a-c
Method A: 1.84 g (0.01 mol) of 2 was dissolved in a warm ethanolic potassium hydroxide solution [prepared by dissolving 0.56 g (0.01 mol) of potassium hydroxide in 50 mL of ethanol]. Then the proper α-haloketone (0.01 mol) was added. The mixture refluxed for 10 minutes; the mixture then stirred at room temperature overnight, then cooled and poured into water. The formed solid product was filtered off and crystallized from ethanol.
Method B: The same reactants of method A were heated in microwave at 500 w and 90 °C for 5 min. The reaction mixture was treated in a similar manner to method A to obtain compounds 10a-c.
2-[6-Hydroxy-4-methyl-2-((2-oxopropyl)thio)]pyridine-3-carboxamide (10a). Obtained as white crystals in 25% yield (Method A) and 55% (Method B); m.p. 241–242 °C. 1H-NMR: 2.00 (s, 3H, CH3), 2.50 (s, 3H, CH3), 4.30 (s, 2H, CH2), 6.15 (pyridine-H), 7.65 (s, 2H, NH2, D2O exchangeable), 11.75 (s, 1H, OH, D2O exchangeable); 13C-NMR: 19.3 (CH3), 28.1 (CH3), 48.3 (CH2), 112.1, 116.2, 146.7, 155.2, 156.0 (sp2 C), 168.4 (CO), 179.3 (CO); IRν: 3295–3446 (broad, NH, OH), 1732, 1678 (2 CO); MS: M+ m/z: 240 (37%). Anal. Calcd for C10H12N2O3S (240.28): C (49.99%), H (5.03%), N (11.66%), S (13.34%). Found: C (50.10%), H (4.90%), N (11.30%), S (13.60%).
2-(2,4-Dioxopentan-3-ylthio)-6-hydroxy-4-methylpyridine-3-carboxamide (10b). Obtained as white crystals in 31% yield (Method A) and 45% yield (Method B); m.p. 311–312 °C. 1H-NMR: 2.20 (s, 3H, CH3), 2.65 (s, 6H, 2CH3), 4.80 (s, 1H, CH), 6.20 (s, 1H, pyridine-H), 7.60 (s, 2H, NH2, D2O exchangeable), 12.00 (s, 1H, OH, D2O exchangeable); 13C-NMR: 19.1 (CH3), 28.5 (CH3), 78.5 (CH), 111.4, 116.8, 146.1, 155.5, 156.4 (sp2 C), 168.0 (CO), 185.3 (CO); IRν: 3288–3433 (broad, NH, OH), 1705, 1678 (2 CO). MS: M+ m/z: 282 (12%). Anal. Calcd for C12H14N2O4S (282.32): C (51.05%), H (5.00%), N (9.92%), S (11.36%). Found: C (51.20%), H (4.90%), N (10.00%), S (11.00%).
2-[6-Hydroxy-4-methyl-2-((2-oxo-2-phenylethyl)thio)]pyridine-3-carboxamide (10c). Obtained as white crystals in 41% yield (Method A) and 63% yield (Method B); m.p. 276–277 °C. 1H-NMR: 2.10 (s, 3H, CH3), 4.95 (s, 2H, CH2), 6.25 (s, 1H, pyridine-H), 7.30 (s, 2H, NH2, D2O exchangeable), 7.56–7.94 (m, 5H, Ar-H), 11.95 (s, 1H, OH, D2O exchangeable); 13C-NMR: 20.1 (CH3), 38.5 (CH2), 112.5, 116.1, 128.5, 128.9, 134.2, 136.0, 146.4, 155.2, 156.8 (sp2 C), 168.3 (CO), 189.0 (CO); IRν: 3471(NH, OH), 1712, 1666 (2 CO). MS: M+ m/z: 302 (21.6%). Anal. Calcd for C15H14N2O3S (302.35): C (59.59%), H (4.67%), N (9.27%), S (10.61%). Found: C (59.30%), H (4.30%), N (9.30%), S (10.60%).
3.8. 7-Bromo-6-methyl-3-phenylpyrido[3,2-f][1,4]thiazepine-5,8(4H,9H)-dione (11)
To a solution of 4c (2.84 g, 0.01 mol) in glacial acetic acid (50 mL), bromine (0.5 mL, 0.01 mole) was added dropwise with stirring at room temperature under sunlight. The mixture was then stirred at water bath for two hours and then diluted with cold water (30 mL). The resultant crude product thus precipitated was collected by filtration, washed with water, dried and crystallized from dilute dimethylformamide to afford 11 as white crystals in 73% yield; m.p. 310–311 °C. 1H-NMR: δ 2.58 (s, 3H, CH3), 7.60–7.86 (m, 6H, Ar-H + thiazepine-H), 13.40 (bs, 2H, NH+OH, D2O exchangeable). 13C-NMR: 12.3 (CH3), 100.2, 108.0, 111.3, 126.9, 127.8, 134.5, 135.6, 137.7, 142.0, 148.4, 152.1 (sp2 C), 165.6 (CO); IRν: 3180-3341 (broad, NH, OH), 1678, (CO). MS: M+ m/z: 365 (64%), 363.0 (66%). Anal. Calcd for C15H11BrN2O2S (363.97): C (49.60%), H (3.05%), Br (22.00%), N (7.71%), S (8.83%). Found: C (50.00%), H (3.30%), Br (21.70%), N (7.52%), S (8.60%).
3.9. 3-Hydroxy-4-methyl-2-(p-substituted phenylazo)-6,7-dihydrothieno[2,3-b]pyridine-6-ones 14a-c
General procedure: To a cold solution of 4 (0.01 mole) in pyridine [50 mL, containing potassium hydroxide, 0.3 g], the arenediazonium chloride (0.01 mL) [prepared by adding concentrated hydrochloric acid (3 mL) to aromatic amine (0.01 mole) at 0 °C and treating the resulting hydrochloride with a cold solution of sodium nitrite 0.69 g (0.01 mole) in water (5 mL)] was added drop-wise with stirring at 0 °C. The coupling mixture was stirred at room temperature for two hours and then diluted with water (30 mL). The crude product thus precipitated was collected by filtration, washed with water, dried and crystallized from dimethylformamide.
3-Hydroxy-4-methyl-2-(phenylazo)-6,7-dihydrothieno[2,3-b]pyridine-6-one (14a). Obtained in 83% yield as an orange crystals; m.p. 320–321 °C. 1H-NMR: 2.44 (s, 3H, CH3), 3.45 (s, 1H, thiazoline-H), 6.20 (s, 1H, pyridine-H), 7.00–7.45 (m, 5H, Ar-H), 11.89 (s, 1H, OH, D2O exchangeable); 13C-NMR: 19.3 (CH3), 75.5 (thiazoline C), 113.5, 115.5, 122.3, 126.4, 129.0, 144.3, 151.1, 155.7, 157.3 (sp2 C), 188.2 (CO); IRν: 3210–2927 (broad, OH), 1639 (CO). MS: M+m/z: 285 (7.3%). Anal. Calcd for C14H11N3O2S (285.32): C (58.93%), H (3.89%), N (14.73%), S (11.24%). Found: C (58.90%), H (4.00%), N (14.50%), S (11.60%).
3-Hydroxy-4-methyl-2-(p-tolylazo)-6,7-dihydrothieno[2,3-b]pyridine-6-one (14b). Obtained in 91% yield as orange crystals; m.p. 338–339 °C. 1H-NMR: δ 2.27 (s, 3H, CH3), 2.43 (s, 3H, CH3), 3.40 (s, 1H, thiazoline-H), 6.14 (s, 1H, pyridine-H), 7.14 (d, 2H, aromatic protons), 7.23 (d, 2H, aromatic protons), 11.61 (s, 1H, OH, D2O exchangeable). 13C-NMR: 18.9 (CH3), 22.3 (CH3), 74.8 (thiazoline c), 113.8, 115.6, 122.9, 130.1, 135.4, 144.8, 147.7, 155.2, 156.6 (sp2 C), 189.0 (CO); IRν: 3200–2900 (broad, OH), 1651 (CO). MS: M+ m/z: 299 (100%). Anal. Calcd for C15H13N3O2S (299.35): C (60.18%), H (4.38%), N (14.04%), S (10.71%). Found: C (60.30%), H (4.30%), N (14.20%), S (10.60%).
3-Hydroxy-4-methyl-2-(p-chlorophenylazo)-6,7-dihydrothieno[2,3-b]pyridine-6-one (14c). Obtained in 85% yield as orange crystals; m.p. 340–341 °C. 1H NMR: δ 2.44 (s, 3H, CH3), 3.45 (s, 1H, thiazoline-H), 6.13 (s, 1H, pyridine-H), 7.27–7.39 (m, 4H, aromatic-H), 13.40 (s, 1H, OH, D2O exchangeable). 13C-NMR: 18.4 (CH3), 73.5 (thiazoline C), 114.3, 115.8, 121.9, 130.8, 135.9, 145.8, 149.7, 155.7, 156.9 (sp2 C), 190.4 (CO); IRν: 3217–2936 (broad, OH), 1651 (CO). MS: M+ m/z: 319 (100%). Anal. Calcd for C14H10ClN3O2S (319.77): C (52.59%), H (3.15%), Cl (11.09%), N (13.14%), S (10.03%). Found: C (52.76%), H (3.30%), Cl (11.40%), N (13.30%), S (10.30%).
3-Hydroxy-4-methyl-2-[(p-methoxyphenyl)azo]-6,7-dihydrothieno[2,3-b]pyridine-6-one (14d). Obtained in 83% yield as orange crystals; m.p. 316–317 °C. 1H-NMR: δ 2.45 (s, 3H, CH3), 3.55 (s, 1H, thiazoline-H), 3.74 (s, 3H, OCH3), 6.13 (s, 1H, pyridine-H), 6.94 (d, 2H, aromatic protons), 7.25 (d, 2H, aromatic protons), 12.84 (s, 1H, OH, D2O exchangeable). 13C-NMR: 18.8 (CH3), 55.6 (OCH3), 73.5 (thiazoline C), 114.1, 115.0, 115.9, 123.8, 143.9, 145.0, 154.7, 156.7, 157.4 (sp2 C), 188.4 (CO); IRν: 3433–2943 (broad, OH), 1655 (CO). MS: M+ m/z: 315 (100%). Anal. Calcd for C15H13N3O3S (315.35): C (57.13%), H (4.16%), N (13.33%), S (10.17%). Found: C (56.90%), H (4.40%), N (13.20%), S (10.10%).






{kind=link}
{kind=link}
{kind=link}
{kind=link}