Derivatives of 10,16-Dihydroxyhexadecanoic Acid Isolated from Tomato (Solanum lycopersicum) as Potential Material for Aliphatic Polyesters

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Isolation of 10,16-Dihydroxyhexadecanoic Acid (1)


{kind=link}
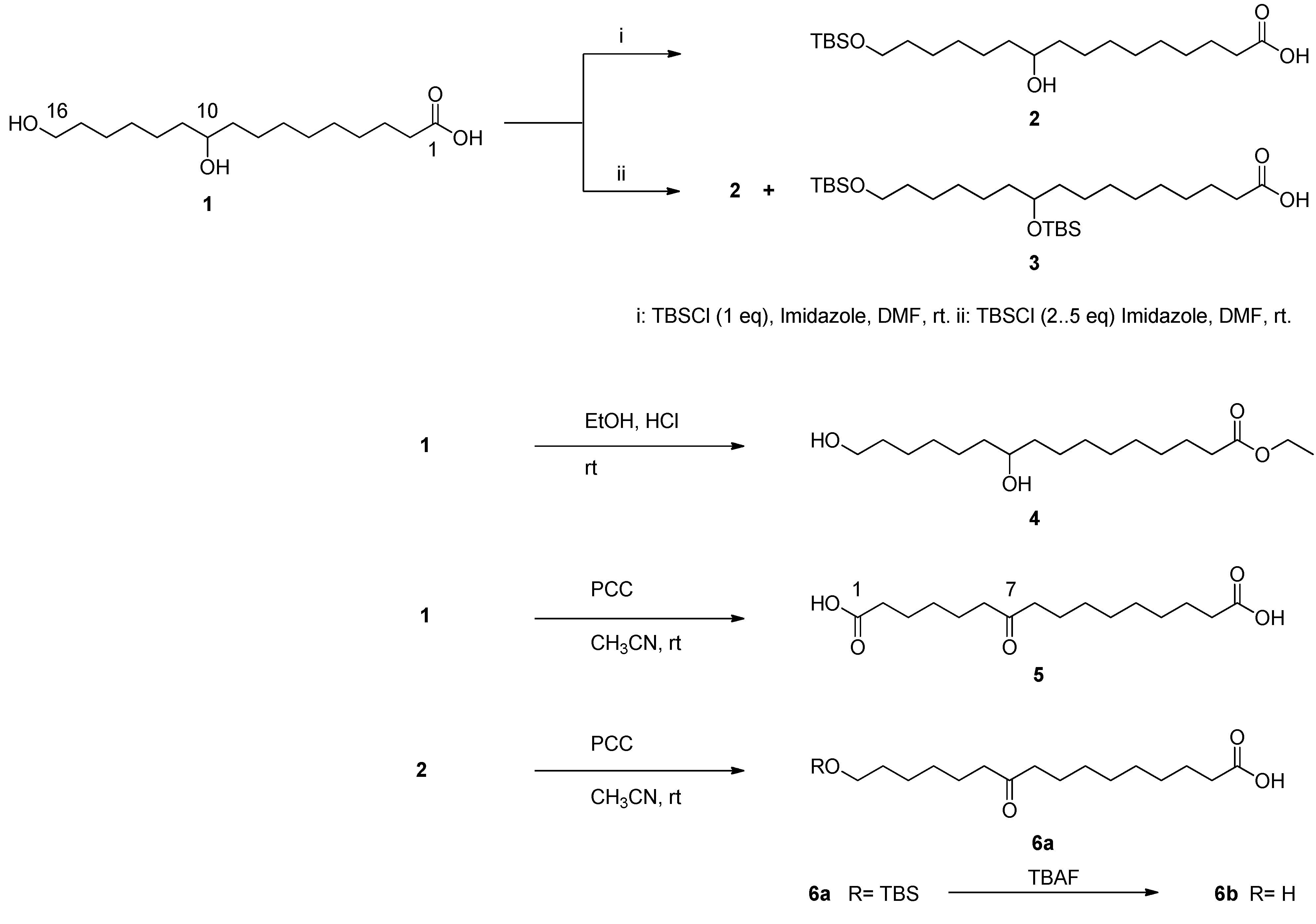{kind=link}
{kind=link}
{kind=link}
| Compounds | MH+ | A | B | C | D | E | F | G |
|---|---|---|---|---|---|---|---|---|
| 1 | 289 | 253(10) | 157(14) 140(−H2O)(9) | 131(26) | 169(-H2O)(100) | --- | 271(1) | 18(4) |
| R1 = H R2 = OH | ||||||||
| R3 = H R4 = H | ||||||||
| 2 | 403 | 385(5) | --- | 245(13) | 187 | --- | 271 | 132(11) |
| R1 = H R2 = OH | 169(−H2O)(11) | 253(−H2O)(8) | ||||||
| R3 = H R4 = TBS | ||||||||
| 3 | 517 | 499(7) | --- | 359(9) | 301(7) | --- | --- | 132(11) |
| R1 = H R2 = OTBS | ||||||||
| R3 = H R4 = TBS | ||||||||
| 4 | 317 | 271(9) | 185(22) | --- | 215(33) | 101(40) | 299(45) | --- |
| R1 = CH2CH3 R2 = OH | ||||||||
| R3 = H R4 = H | ||||||||
| 5 | 300 | ---- | 157(79) | --- | 185(62) | ---- | --- | ---- |
| R1 = H R2 = (=O) | ||||||||
| R3 = (=O) R4 = H | ||||||||
| 6b | --- | 269(34) | --- | --- | 185(28) | --- | --- | 251(35) b |
| R1 = H R2 = (=O) | ||||||||
| R3 = H R4 = H |
2.2. Preparation OF 7-Oxohexadecanedioic ACID (5)
2.3. Preparation OF 16-Hydroxy-10-Oxo-Hexadecanoic ACID (6b)
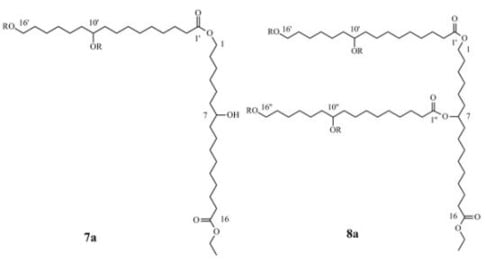
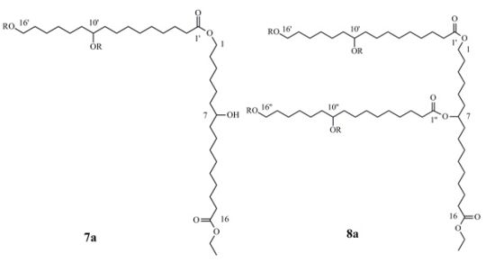
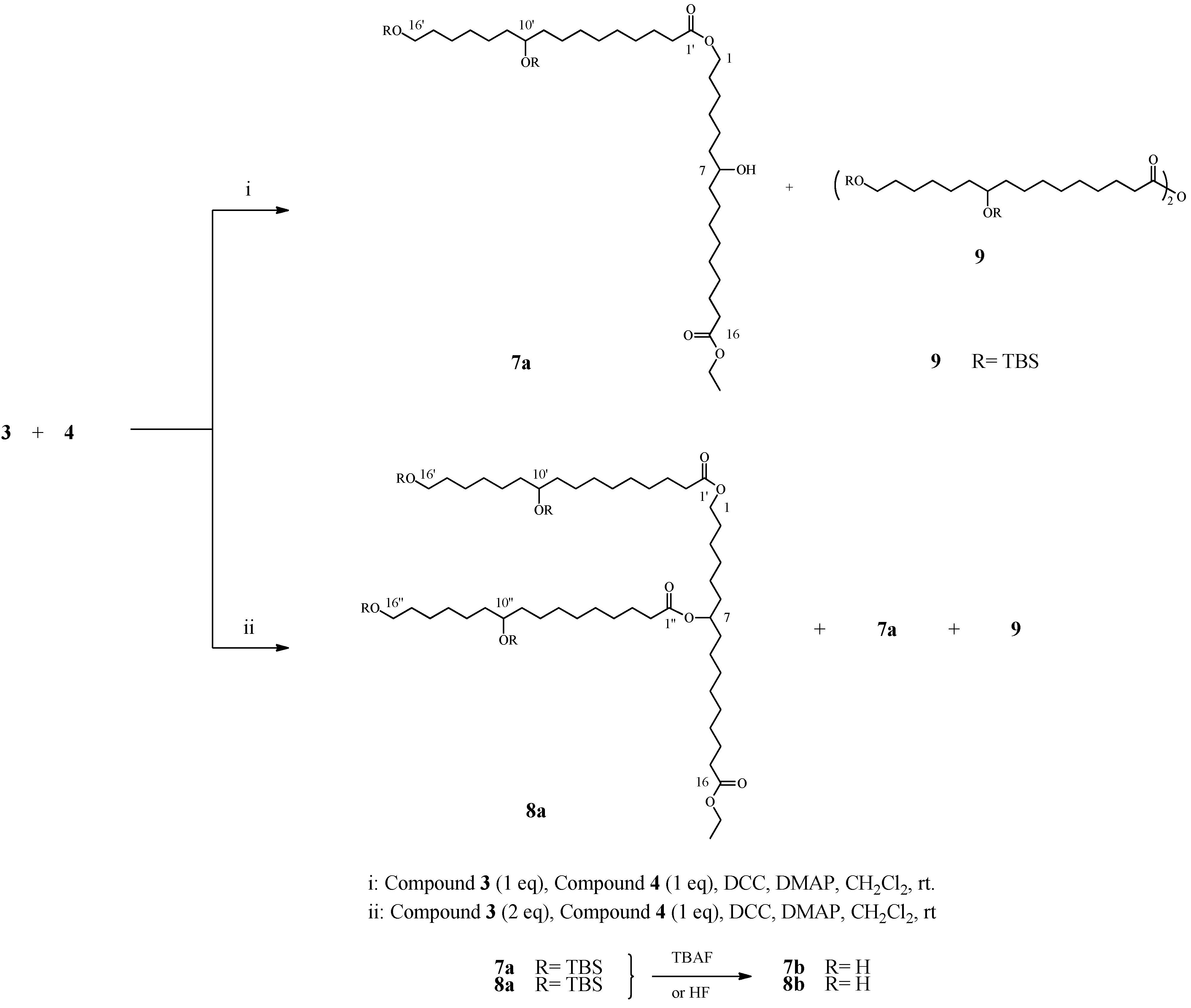
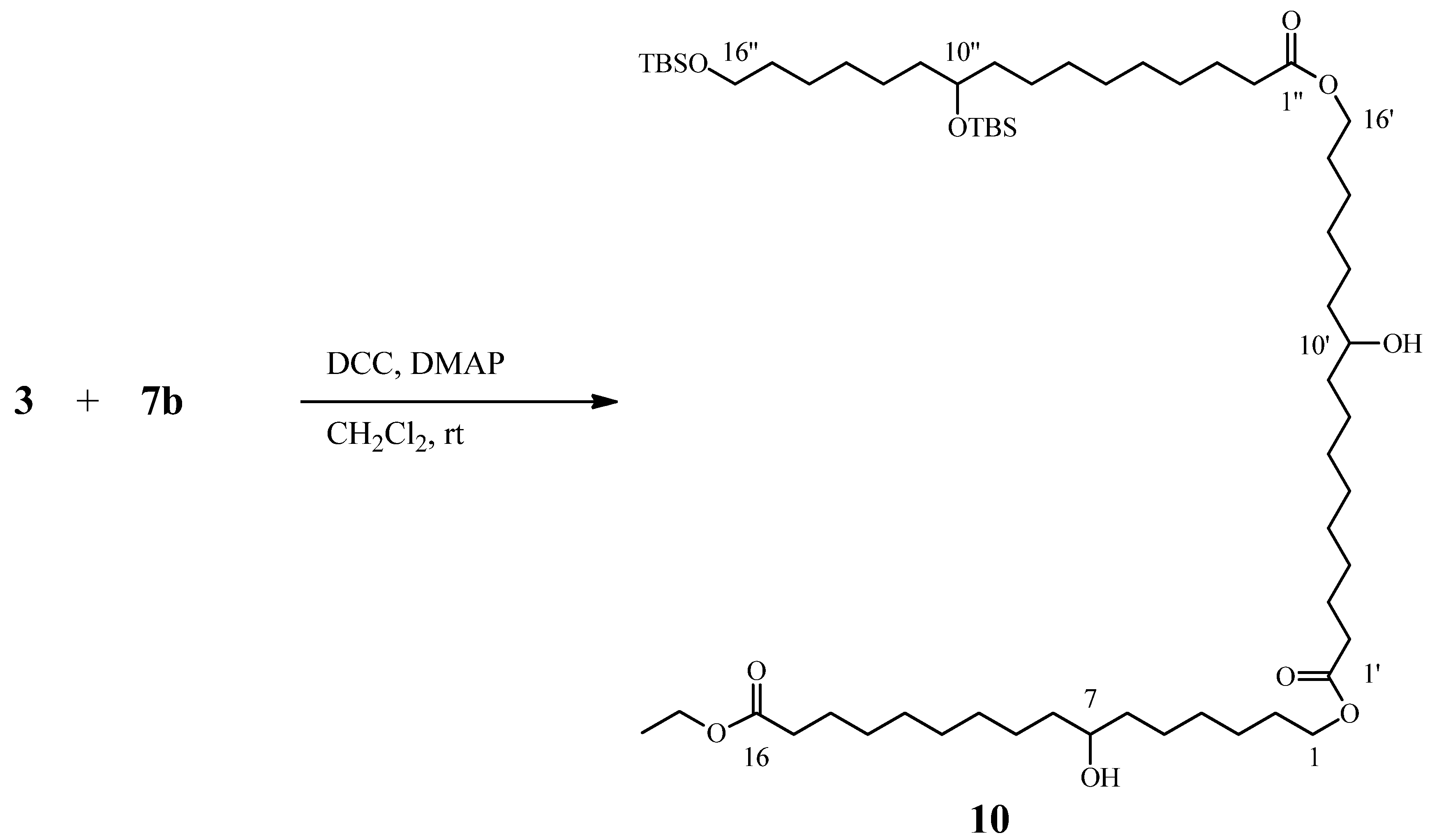
2.4. Preparation and Characterization OF Dimer 7 and Trimer 8


2.5. Preparation of Linear Trimer

3. Experimental
3.1. General
3.2. Isolation of 10,16-Dihydroxypalmitic Acid (10,16-DHPA, 1)
3.3. Synthesis
3.3.1. Preparation of 10-hydroxy-16-OTBS-PA (2) and 10,16-di-OTBS-PA (3)
3.3.2. Preparation of ethyl-10,16-dihydroxy hexadecanoate (4)
3.3.3. Oxidation of 10,16-DHPA (1) and compound 2 with pyridinium chlorochromate (PCC)
3.3.4. Dimerization of 10,16-DHPA derivatives
3.3.5. Branched trimer of 10,16-DHPA derivatives
3.3.6. Linear trimer of 10,16-DHPA derivatives
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Narayan, N. Drivers for biodegradable/compostable plastics and role of composting in waste management and sustainable agriculture. Orbit J. 2001, 1, 1–9. [Google Scholar]
- Gross, R.A.; Kalra, B. Biodegradable polymers for the environment. Science 2002, 297, 803–807. [Google Scholar] [CrossRef]
- van der Walle, G.A.; de Koning, G.J.; Weusthuis, R.A.; Eggink, G. Properties, modifications and applications of biopolyesters. Adv. Biochem. Eng. Biotechnol. 2002, 71, 263–291. [Google Scholar]
- Bordes, P.; Pollet, E.; Averous, L. Nano-biocomposites: Biodegradable polyester/nanoclay systems. Prog.Polymer Sci. 2009, 34, 125–155. [Google Scholar] [CrossRef]
- Mahapatro, A.; Kalra, B.; Kumar, A.; Gross, R.A. Lipase-catalyzed polycondensations: Effect of substrates and solvent on chain formation, dispersity, and end-group structure. Biomacromolecules 2003, 4, 544–551. [Google Scholar] [CrossRef]
- Moon, S.I.; Lee, C.W.; Taniguchi, I.; Miyamoto, M.; Kimura, Y. Melt/Solid polycondensation of L-lactic acid: An alternative route to poly(L-lactic acid) with high molecular weight. Polymer 2001, 42, 5059–5062. [Google Scholar]
- Yang, Y.; Lu, W.; Zhang, X.; Xie, W.; Cai, M.; Gross, R.A. Two-step biocatalytic route to biobased functional polyesters from ω-carboxy fatty acids and diols. Biomacromolecules 2010, 11, 259–268. [Google Scholar] [CrossRef]
- Uyama, H.; Takeya, S.; Kobayashi, S. Synthesis of polyesters by enzymic ring-opening copolymerization using lipase catalyst. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 1993, 69, 203–207. [Google Scholar] [CrossRef]
- Rehm, B.H. Polyester synthases: Natural catalysts for plastics. Biochem.J. 2003, 376, 15–33. [Google Scholar] [CrossRef]
- Bernards, M.A.; Lewis, N.G. The macromolecular aromatic domain in suberized tissue: A changing paradigm. Phytochemistry 1998, 47, 915–933. [Google Scholar]
- Gerard, H.C.; Osman, S.F.; Fett, W.F.; Moreau, R.A. Separation, identification and quantification of monomers from cutin polymers by high performance liquid chromatography and evaporative light scattering detection. Phytochem.Anal. 1992, 3, 139–144. [Google Scholar] [CrossRef]
- Kolattukudy, P.E. Polyesters in higher plants. Adv. Biochem. Eng Biotechnol. 2001, 71, 1–49. [Google Scholar]
- Fang, X.; Qiu, F.; Yan, B.; Wang, H.; Mort, A.J.; Stark, R.E. NMR studies of molecular structure in fruit cuticle polyesters. Phytochemistry 2001, 57, 1035–1042. [Google Scholar]
- Kallio, H.; Nieminen, R.; Tuomasjukka, S.; Hakala, M. Cutin composition of five finnish berries. J. Agric. Food Chem. 2006, 54, 457–462. [Google Scholar] [CrossRef]
- Osman, S.F.; Irwin, P.; Fett, W.F.; O’Connor, J.V.; Parris, N. Preparation, isolation, and characterization of cutin monomers and oligomers from tomato peels. J. Agric. Food Chem. 1999, 47, 799–802. [Google Scholar] [CrossRef]
- Ray, A.K.; Stark, R.E. Isolation and molecular structure of an oligomer produced enzymatically from the cuticle of lime fruit. Phytochemistry 1998, 48, 1313–1320. [Google Scholar]
- Suh, M.C.; Samuels, A.L.; Jetter, R.; Kunst, L.; Pollard, M.; Ohlrogge, J.; Beisson, F. Cuticular lipid composition, surface structure, and gene expression in Arabidopsis stem epidermis. Plant Phys. 2005, 139, 1649–1665. [Google Scholar] [CrossRef]
- Mahapatro, A.; Kumar, A.; Gross, R.A. Mild, solvent-free omega-hydroxy acid polycondensations catalyzed by candida antarctica lipase B. Biomacromolecules 2004, 5, 62–68. [Google Scholar] [CrossRef]
- Benitez, J.J.; Heredia-Guerrero, J.A.; Serrano, F.M.; Heredia, A. The role of hydroxyl groups in the self-assembly of long chain alkylhydroxyl carboxylic acids on Mica. J. Phys. Chem. C 2008, 112, 16968–16972. [Google Scholar] [CrossRef]
- Heredia-Guerrero, J.A.; Heredia, A.; García-Segura, R.; Benitez, J.J. Synthesis and characterization of a plant cutin mimetic polymer. Polymer 2009, 50, 5633–5637. [Google Scholar]
- Douliez, J.P. Cutin and suberin monomers are membrane perturbants. J. Colloid Interface Sci. 2004, 271, 507–510. [Google Scholar] [CrossRef]
- Osman, S.F.; Gerard, H.C.; Fett, W.F.; Moreau, R.A.; Dudley, R.L. Method for the production and characterization of tomato cutin oligomers. J. Agric. Food Chem. 2002, 43, 2134–2137. [Google Scholar]
- Stark, R.E.; Yan, B.; Ray, A.K.; Chen, Z.; Fang, X.; Garbow, J.R. NMR studies of structure and dynamics in fruit cuticle polyesters. Solid State Nucl.Magn. Reson. 2000, 16, 37–45. [Google Scholar] [CrossRef]
- Croteau, R.; Kolattukudy, P.E. Biosynthesis of hydroxyfatty acid polymers. Enzymatic synthesis of cutin from monomer acids by cell-free preparations from the epidermis of Vicia faba leaves. Biochemistry 1974, 13, 3193–3202. [Google Scholar] [CrossRef]
- Tian, S.; Fang, X.; Wang, W.; Yu, B.; Cheng, X.; Qiu, F.; Mort, A.J.; Stark, R.E. Isolation and identification of oligomers from partial degradation of lime fruit cutin. J. Agric. Food Chem. 2008, 56, 10318–10325. [Google Scholar]
- Matic, M. The chemistry of plant cuticles: A study of cutin from Agave americana L. Biochem. J. 1956, 63, 168–176. [Google Scholar]
- Ahmed, A.; Crawford, T.; Gould, S.; Ha, Y.S.; Hollrah, M.; Noor-E-Ain.; Dickman, M.B.; Dussault, P.H. Synthesis of (R)- and (S)-10,16-dihydroxyhexadecanoic acid: Cutin stereochemistry and fungal activation. Phytochemistry 2003, 63, 47–52. [Google Scholar]
- Benitez, J.J.; Garcia-Segura, R.; Heredia, A. Plant biopolyester cutin: A tough way to its chemical synthesis. Biochim.Biophys. Acta 2004, 1674, 1–3. [Google Scholar]
- Benitez, J.J.; Heredia-Guerrero, J.A.; Heredia, A. Self-Assembly of carboxylic acids and hydroxyl derivatives on Mica. A Qualitative AFM Study. J. Phys. Chem. C 2007, 111, 9465–9470. [Google Scholar]
- Deshmuk, A.P.; Simpson, A.J.; Hatcher, P.G. Evidence for cross-linking in tomato cutin using HR-MAS NMR spectroscopy. Phytochemistry 2003, 64, 1163–1170. [Google Scholar]
- Graca, J.; Lamosa, P. Linear and branched poly(w-hydroxyacid) ester in plant cutins. J. Agric. Food Chem. 2010, 58, 9666–9674. [Google Scholar] [CrossRef]
- Samples Availability: Samples of the compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arrieta-Baez, D.; Cruz-Carrillo, M.; Gómez-Patiño, M.B.; Zepeda-Vallejo, L.G. Derivatives of 10,16-Dihydroxyhexadecanoic Acid Isolated from Tomato (Solanum lycopersicum) as Potential Material for Aliphatic Polyesters. Molecules 2011, 16, 4923-4936. https://doi.org/10.3390/molecules16064923
Arrieta-Baez D, Cruz-Carrillo M, Gómez-Patiño MB, Zepeda-Vallejo LG. Derivatives of 10,16-Dihydroxyhexadecanoic Acid Isolated from Tomato (Solanum lycopersicum) as Potential Material for Aliphatic Polyesters. Molecules. 2011; 16(6):4923-4936. https://doi.org/10.3390/molecules16064923
Chicago/Turabian StyleArrieta-Baez, Daniel, Miguel Cruz-Carrillo, Mayra Beatriz Gómez-Patiño, and L. Gerardo Zepeda-Vallejo. 2011. "Derivatives of 10,16-Dihydroxyhexadecanoic Acid Isolated from Tomato (Solanum lycopersicum) as Potential Material for Aliphatic Polyesters" Molecules 16, no. 6: 4923-4936. https://doi.org/10.3390/molecules16064923
APA StyleArrieta-Baez, D., Cruz-Carrillo, M., Gómez-Patiño, M. B., & Zepeda-Vallejo, L. G. (2011). Derivatives of 10,16-Dihydroxyhexadecanoic Acid Isolated from Tomato (Solanum lycopersicum) as Potential Material for Aliphatic Polyesters. Molecules, 16(6), 4923-4936. https://doi.org/10.3390/molecules16064923