Enantioselective Addition of Allyltin Reagents to Amino Aldehydes Catalyzed with Bis(oxazolinyl)phenylrhodium(III) Aqua Complexes

Abstract
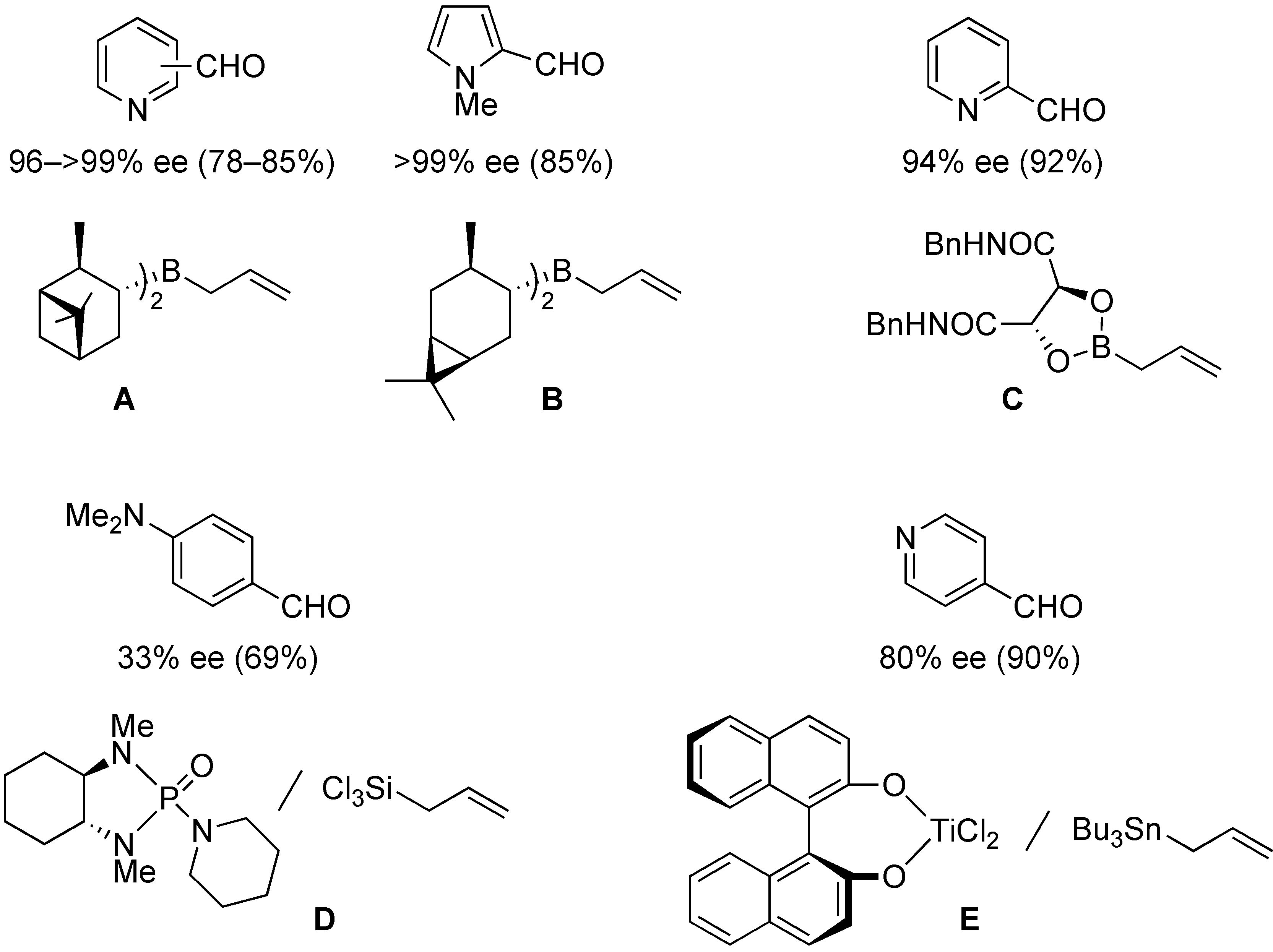
:1. Introduction


2. Results and Discussion
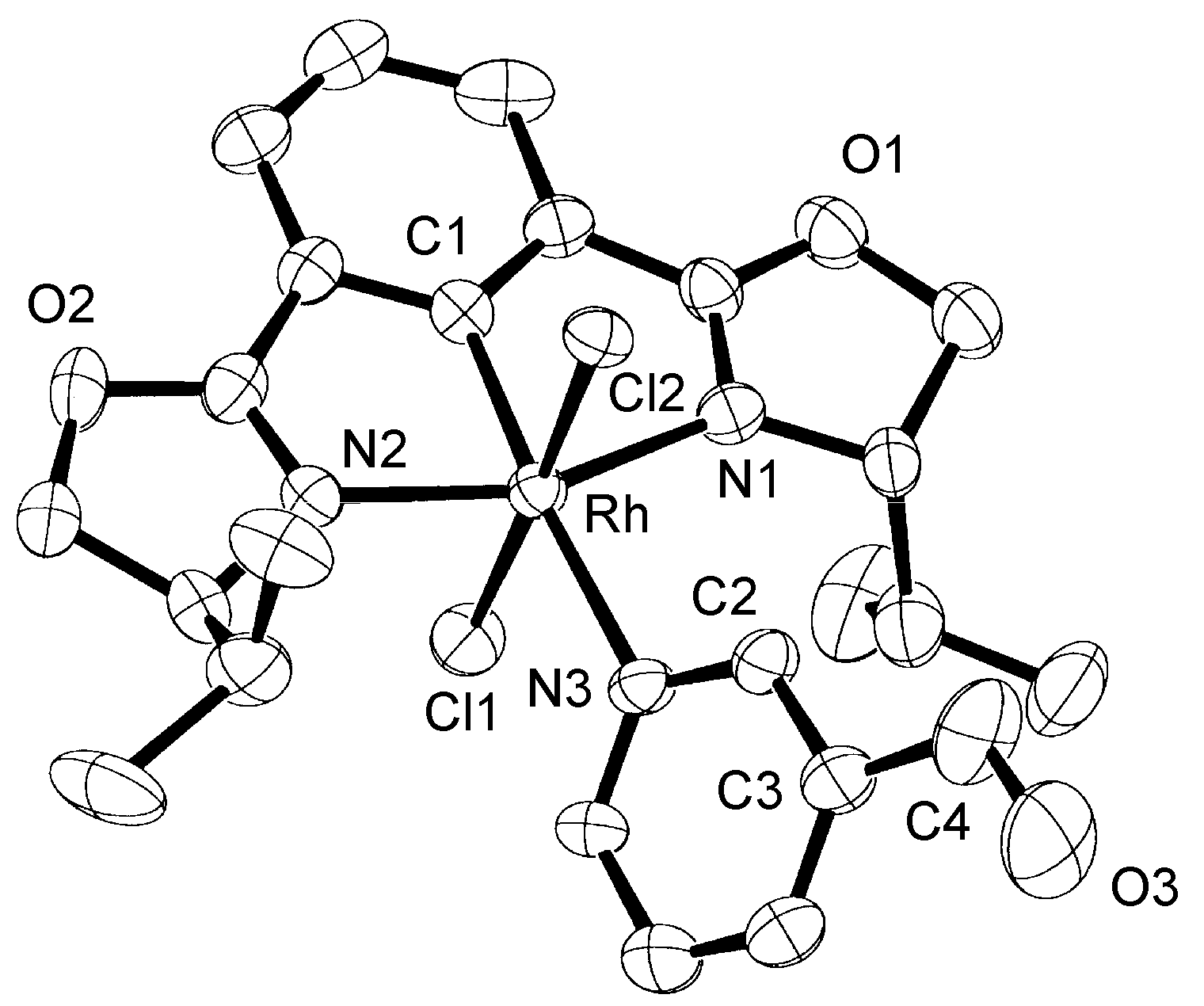
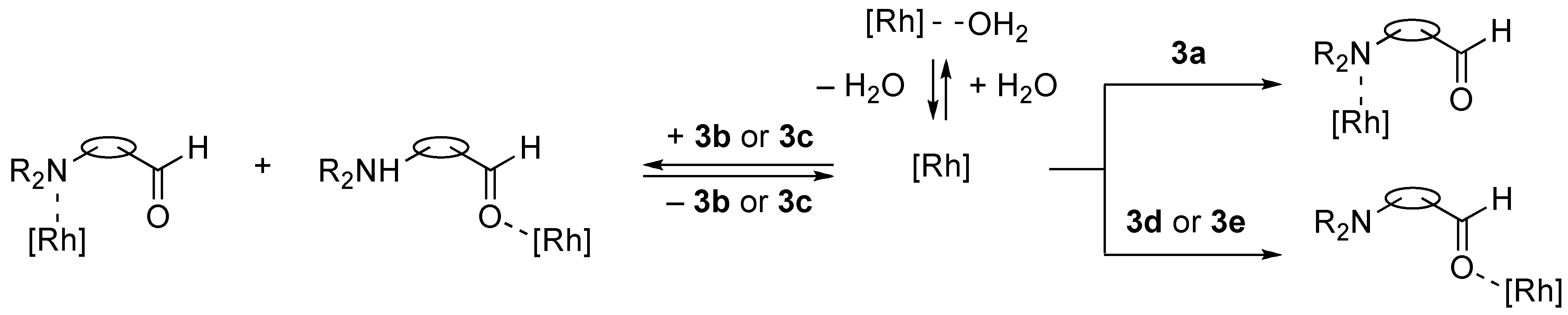
2.1. NMR Studies, Isolation, and X-ray Analysis of Phebox-Rh(III)–Amino Aldehyde Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | δ (ppm) a | Δ (ppm) b | ||
|---|---|---|---|---|
| 3 | i-Pr-1 and 3 | |||
| 1 |  | H2: 9.08 | H2: 10.30 | +1.22 |
| H6: 8.85 | H6: 10.08 | +1.23 | ||
| Hf: 10.12 | Hf: 10.27 | +0.15 | ||
| Cf: 190.8 | Cf: 189.8 | –1.0 | ||
| 2 | 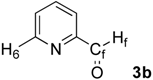 | H6: 8.77 | H6: 9.11 (br) | +0.34 |
| Hf: 10.07 | Hf: 10.34 (br) | +0.27 | ||
| 3 | 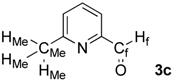 | HMe: 2.66 | HMe: 2.66 | 0.00 |
| Hf: 10.04 | Hf: 10.06 | +0.02 | ||
| CMe: 24.5 | CMe: 24.5 | 0.0 | ||
| Cf: 193.1 | Cf: 194.1 | +1.0 | ||
| 4 | 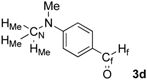 | HMe: 3.09 | HMe: 3.11 | +0.02 |
| Hf: 9.74 | Hf: 9.92 | +0.18 | ||
| CN: 40.2 | CN: 40.2 | 0.0 | ||
| Cf: 190.4 | Cf: 207.2 | +16.8 | ||
| 5 | 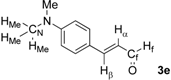 | Hα: 6.54 | Hα: 6.81 | +0.27 |
| Hβ: 7.38 | Hβ: 7.52 | +0.14 | ||
| HMe: 3.05 | HMe: 3.07 | +0.02 | ||
| Hf: 9.09 | Hf: 9.86 | +0.77 | ||
| CN: 40.2 | CN: 40.2 | 0.0 | ||
| Cf: 193.8 | Cf: 196.7 | +2.9 | ||


| Rh–C1 | 1.93(1) [1.89(1)] | Rh–N1 | 2.05(1) [2.06(1)] |
| Rh–Cl1 | 2.340(4) [2.334(4)] | Rh–N2 | 2.05(1) [2.09(1)] |
| Rh–Cl2 | 2.334(4) [2.351(4)] | Rh–N3 | 2.21(1) [2.27(1)] |
| C4–O3 | 1.25(3) [1.27(4)] | ||
| C1–Rh–N3 | 175.2(6) [178.0(5)] | N1–Rh–N3–C2 | 54(1) [90(1)] |
| Cl1–Rh–Cl2 | 178.0(2) [177.2(2)] | O3–C4–C3–C2 | −175(2) [19(3)] |
| N1–Rh–N2 | 158.4(5) [157.6(5)] |
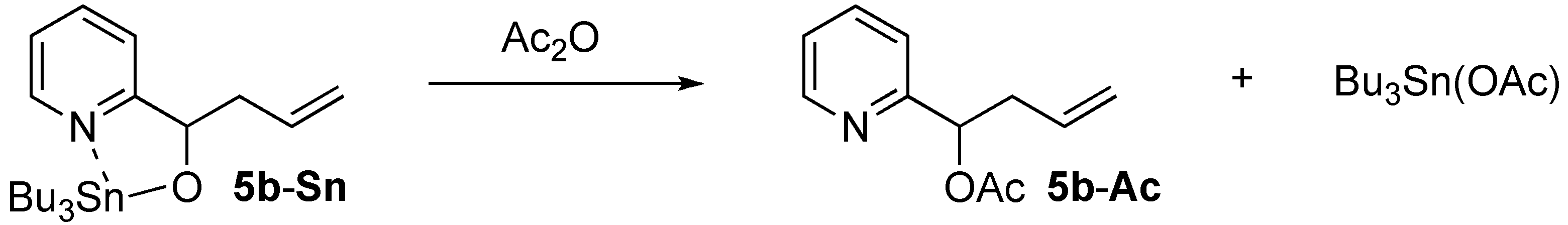
2.2. Phebox-Rh(III)-Catalyzed Enantioselective Addition of Allyltributyltin to Amino Aldehydes

| Entry | Aldehyde | Catalyst | Product | % Yield | % eeb (config.) c |
|---|---|---|---|---|---|
| 1 | 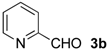 | i-Pr-1 | 5b | 14 | 42 (S) |
| 2 d | i-Pr-1 | 5b-Ac | 99 | 53 (S) | |
| 3 d | Me- 1 | 5b-Ac | 99 | 56 (S) | |
| 4 d | Ph- 1 | 5b-Ac | 45 | 21 (R) | |
| 5 d | i-Pr-2 | 5b-Ac | 81 | 59 (S) | |
| 6 d | Me- 2 | 5b-Ac | 85 | 59 (S) | |
| 7 | 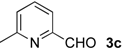 | i-Pr-1 | 5c | 94 | 69 (S)e |
| 8 | Ph- 1 | 5c | 89 | 75 (S)e | |
| 9 | Ph- 2 | 5c | 97 | 84 (S)e | |
| 10 | 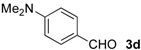 | Me- 1 | 5d | 80 | 84 (S) |
| 11 | s-Bu-1 | 5d | 67 | 81 (S) | |
| 12 | Me- 2 | 5d | 42 | 72 (S) | |
| 13 | 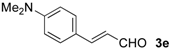 | Bn- 1 | 5e | 80 | 81 (S)e |
| 14 | s-Bu-1 | 5e | 61 | 80 (S)e | |
| 15 | Bn- 2 | 5e | 44 | 88 (S)e |
2.3. Phebox-Rh(III)-Catalyzed Enantioselective Addition of Methallyltributyltin to Amino Aldehydes
| Entry | Aldehyde | Catalyst | Product | % Yield | % ee b (config.) c |
|---|---|---|---|---|---|
| 1 d | 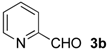 | Bn- 1 | 6b-Ac | 79 | 15 (S) |
| 2 d | Me- 1 | 6b-Ac | 76 | 41 (S) | |
| 3 d | Ph- 1 | 6b-Ac | 18 | 24 (R) | |
| 4 d | s-Bu-1 | 6b-Ac | 52 | <2 (–) | |
| 5 d | Me- 2 | 6b-Ac | 48 | 45 (S) | |
| 6 d | s-Bu-2 | 6b-Ac | 22 | 51 (S) | |
| 7 | 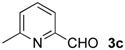 | Me- 1 | 6c | 60 | 45 (S)e |
| 8 | s-Bu-1 | 6c | 36 | 11 (S)e | |
| 9 | Me- 2 | 6c | 21 | 10 (S)e | |
| 10 | s-Bu-2 | 6c | 26 | 26 (S)e | |
| 11 | 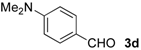 | i-Pr-1 | 6d | 84 | 85 (S)e |
| 12 | Bn- 1 | 6d | 79 | 90 (S)e | |
| 13 | s-Bu-1 | 6d | 68 | 87 (S)e | |
| 14 | Bn- 2 | 6d | 52 | 63 (S)e | |
| 15 | 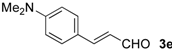 | i-Pr-1 | 6e | 52 | 80 (S)e |
| 16 | s-Bu-1 | 6e | 74 | 84 (S)e | |
| 17 | s-Bu-2 | 6e | 20 | 94 (S)e |
3. Experimental
3.1. General
3.2. General Procedure for the Synthesis of (Phebox)RhBr2(H2O) Complexes
3.3. General Procedure for the Catalytic Enantioselective Addition of Allyl- or Methallyltributyltin to Aldehydes Catalyzed with (Phebox)RhX2(H2O) Complexes
3.4. Synthesis and X-ray Analysis of (i-Pr-Phebox)RhCl2 (κ-3a)
| Empirical Formula | C48H58N6O7Cl4Rh2 | Temperature | 23.0 °C | |
| Formula Weight | 1178.65 | Scan type | ω -2 θ | |
| Crystal Dimensions | 0.15 × 0.5 × 0.5 mm | Scan Width | 94 | |
| 3 tan θ deg | ||||
| Crystal System | monoclinic | 2θmax | 55.0 deg | |
| Lattice Type | C-centered | No. of Reflection | Total: 6787 | |
| Lattice Parameters: a | 18.307(4) Å | measured | ||
| b | 14.886(5) Å | No. of Unique data | 6581 (Rint = 0.018) | |
| c | 21.056(4) Å | Structure Solution | Direct methods | |
| β | 106.55(2) deg | Refinement | Full-matrix | |
| Volume | 5500(2) Å3 | least squares | ||
| Space Group | C2 (#5) | No. of Observations | 5306 (I>3σ(I)) | |
| Z value | 4 | No. of Variables | 598 | |
| Dcalcd | 1.423 g/cm3 | Reflection/Parameter | 8.87 | |
| F(000) | 2408.00 | Ratio | ||
| μ(Mo Kα) | 8.44 cm−1 | Residuals: R; Rw | 0.058; 0.077 | |
| λ | 0.71069 Å | |||
4. Conclusions
Acknowledgements
References and Notes
- Roush, W.R. Allyl Organometallics. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Heathcock, C.H., Eds.; Pergamon Press: Oxford, UK, 1991; Volume 2, pp. 1–55. [Google Scholar]
- Yamamoto, Y.; Asao, N. Selective Reactions using Allylic Metals. Chem. Rev. 1993, 93, 2207–2293. [Google Scholar] [CrossRef]
- Yanagisawa, A. Allylation of Carbonyl Groups. In Comprehensive Asymmetric Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Heidelberg, Germany, 1999; Volume 2, pp. 965–982. [Google Scholar]
- Racherla, U.S.; Liao, Y.; Brown, C.H. Chiral Synthesis via Organoboranes. 36. Exceptionally Enantioselective Allylboration of Representative Heterocyclic Aldehydes at −100 °C under Salt-Free Conditions. J. Org. Chem. 1992, 57, 6614–6617. [Google Scholar]
- Chen, W.; Liu, Y.; Chen, Z. A Highly Efficient and Practical New Allylboronate Tetramide for the Asymmetric Allylboration of Achiral Aldehydes. Eur. J. Org. Chem. 2005, 1665–1668. [Google Scholar]
- Denmark, S.E.; Coe, D.M.; Pratt, N.E.; Griedel, B.D. Asymmetric Allylation of Aldehydes with Chiral Lewis Bases. J. Org. Chem. 1994, 59, 6161–6163. [Google Scholar] [CrossRef]
- Costa, A.L.; Piazza, M.G.; Tagliavini, E.; Trombini, C.; Umani-Ronchi, A. Catalytic Asymmetric Synthesis of Homoallylic Alcohols. J. Am. Chem. Soc. 1993, 115, 7001–7002. [Google Scholar]
- Motoyama, Y.; Nishiyama, H. Asymmetric Reactions with Chiral Bis(oxazolinyl)phenyl-Rh, -Pt, and -Pd Complexes and Their Lewis Acid Activity. In Latest Frontiers of Organic Synthesis; Kobayashi, Y., Ed.; Research Signpost: Kerala, India, 2002; pp. 1–24. [Google Scholar]
- Nishiyama, H. Synthesis and Use of Bisoxazolinyl-phenyl Pincers. Chem. Soc. Rev. 2007, 36, 1133–1141. [Google Scholar] [CrossRef]
- Stark, M.A.; Richards, C.J. Synthesis and Application of Cationic 2,6-Bis(2-oxazolilyl)phenylpalladium(II) Complexes. Tetrahedron Lett. 1997, 38, 5881–5884. [Google Scholar] [CrossRef]
- Denmark, S.E.; Stavenger, R.A.; Faucher, A.-M.; Edwards, J.P. Cyclopropanation with Diazomethane and Bis(oxazoline)palladium(II) Complexes. J. Org. Chem. 1997, 62, 3375–3389. [Google Scholar] [CrossRef]
- Stol, M.; Snelders, D.J.M.; de Pater, J.J.M.; van Klink, G.P.M.; Kooijman, H.; Spek, A.L.; van Koten, G. Organometallics 2005, 24, 743–749.
- Kimura, T.; Uozumi, Y. Synthesis of [2,6-Bis(2-oxazolinyl)phenyl]palladium Complexes via the Ligand Introduction Route. Organometallics 2008, 27, 5159–5162. [Google Scholar] [CrossRef]
- Chuchuryukin, A.V.; Huang, R.; Lutz, M.; Chadwick, J.C.; Spek, A.L.; van Koten, G. NCN-Pincer Metal Complexes (Ti, Cr, V, Zr, Hf, and Nb) of the Phebox Ligand (S,S)-2,6-Bis(4’-isopropyl-2’-oxazolinyl)phenyl. Organometallics 2011, 30, 2819–2830. [Google Scholar] [CrossRef]
- Motoyama, Y.; Narusawa, H.; Nishiyama, H. Chiral Bis(oxazolinyl)phenylrhodium(III) Complexes as Lewis Acid Catalysts for Enantioselective Allylation of Aldehydes. Chem. Commun. 1999, 131–132. [Google Scholar]
- Motoyama, Y.; Okano, M.; Narusawa, H.; Makihara, N.; Aoki, K.; Nishiyama, H. Bis(oxazolinyl)phenylrhodium(III) Aqua Complexes: Synthesis, Structure, Enantioselective Allylation of Aldehydes and Mechanistic Studies. Organometallics 2001, 20, 1580–1591. [Google Scholar] [CrossRef]
- Motoyama, Y.; Nishiyama, H. Bis(oxazolinyl)phenylrhodium(III) Aqua Complex: Efficiency in Enantioselective Addition of Methallyltributyltin to Aldehydes under Aerobic Conditions. Synlett 2003, 1883–1885. [Google Scholar] [CrossRef]
- Motoyama, Y.; Koga, Y.; Nishiyama, H. Asymmetric Hetero Diels-Alder Reaction of Danishefsky’s Dienes and Glyoxylates with Chiral Bis(oxazolinyl)phenylrhodium(III) Aqua Complexes, and Its Mechanistic Studies. Tetrahedron 2001, 57, 853–860. [Google Scholar] [CrossRef]
- Motoyama, Y.; Koga, Y.; Kobayashi, K.; Aoki, K.; Nishiyama, H. Novel Asymmetric Michael Addition of α-Cyanopropionates to Acrolein by the Use of a Bis(oxazolinyl)phenylstannane-Derived Rhodium(III) Complex as a Chiral Lewis Acid Catalyst. Chem. Eur. J. 2002, 8, 2968–2975. [Google Scholar] [CrossRef]
- Furukawa, J.; Kobayashi, E.; Nagata, S.; Moritani, T. C-13 NMR Spectroscopy of Acrylic Monomer and Lewis Acid Complexes. J. Polym. Sci. A. Polym. Chem. 1974, 12, 1799–1807. [Google Scholar]
- Kuran, W.; Pasynkiewicz, S.; Florjanczyk, K.; Lusztyk, E. Vinyl Monomer Complexes in Copolymerization Processes – Studies on Complexes of Acrylonitrile and Methyl Methacrylate with Lewis Acids. Macromol. Chem. 1976, 177, 2627–2635. [Google Scholar] [CrossRef]
- Childs, R.F.; Mulholland, D.L.; Nixon, A. The Lewis Acid Complexes of α,β-Unsaturated Carbonyl and Nitrile Compounds 1. A Nuclear Magnetic-Resonance Study. Can. J. Chem. 1982, 60, 801–808. [Google Scholar] [CrossRef]
- Uenishi, J.; Hiraoka, T.; Hata, S.; Nishiwaki, K.; Yonemitsu, O.; Nakamura, K.; Tsukube, H. Chiral Pyridines: Optical Resolution of 1-(2-Pyridyl)- and 1-[6-(2,2’-Bipyridyl)]ethanols by Lipase-Catalyzed Enantioselective Acetylation. J. Org. Chem. 1998, 63, 2481–2487. [Google Scholar] [CrossRef]
- Motoyama, Y.; Kawakami, H.; Shimozono, K.; Aoki, K.; Nishiyama, H. Synthesis and X-ray Crystal Structures of Bis(oxazolinyl)phenyl-Derived Chiral Palladium(II) and Platinum(II) and –(IV) Complexes and Their Use in the Catalytic Asymmetric Aldol-Type Condensation of Isocyanides and Aldehydes. Orgnometallics 2002, 21, 3408–3414. [Google Scholar] [CrossRef]
- van der Ent, A.; Onderdelinden, A.C. Chlorobis(cyclooctene)rhodium(I) and Chlorobis(cyclooctene)iridium(I) Complexes. Inorg. Synth. 1990, 28, 90–92. [Google Scholar] [CrossRef]
- Still, W.C. Stannylation/Destannylation. Preparation of α-Alkoxy Organolithium Reagents and Synthesis of Dendrolasin via a Carbonyl Carbanion Equivalent. J. Am. Chem. Soc. 1978, 100, 1481–1487. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.; Polidori, G.; Camalli, M. SIR92: A new tool for crystal structure determination and refinement. J. Appl. Crystalogr. 1994, 27, 435. [Google Scholar]
- Beurskens, P.T.; Admiraal, G.; Beurskens, G.; Bosman, W.P.; Garcia-Granda, S.; Gould, R.O.; Smits, J.M.M.; Smykalla, C. DIRDIF92: The DIRDIF Program System; Technical Report of the Crystallography Laboratory, University of Nijmegen: The Netherlands, 1992.
- Cromer, D.T.; Waber, J.T. International Tables for X-ray Crystallography; Kynoch Press: Birmingham, UK, 1974; Volume 4. [Google Scholar]
- teXane: Crystal Structure Analysis Package, Molecular Structure Corporation: The Woodlands, TX, USA, 1985 & 1992.
- Sample availability: Samples of the complexes 1 and 2 are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Motoyama, Y.; Sakakura, T.; Takemoto, T.; Shimozono, K.; Aoki, K.; Nishiyama, H. Enantioselective Addition of Allyltin Reagents to Amino Aldehydes Catalyzed with Bis(oxazolinyl)phenylrhodium(III) Aqua Complexes. Molecules 2011, 16, 5387-5401. https://doi.org/10.3390/molecules16075387
Motoyama Y, Sakakura T, Takemoto T, Shimozono K, Aoki K, Nishiyama H. Enantioselective Addition of Allyltin Reagents to Amino Aldehydes Catalyzed with Bis(oxazolinyl)phenylrhodium(III) Aqua Complexes. Molecules. 2011; 16(7):5387-5401. https://doi.org/10.3390/molecules16075387
Chicago/Turabian StyleMotoyama, Yukihiro, Takatoshi Sakakura, Toshihide Takemoto, Kayoko Shimozono, Katsuyuki Aoki, and Hisao Nishiyama. 2011. "Enantioselective Addition of Allyltin Reagents to Amino Aldehydes Catalyzed with Bis(oxazolinyl)phenylrhodium(III) Aqua Complexes" Molecules 16, no. 7: 5387-5401. https://doi.org/10.3390/molecules16075387
APA StyleMotoyama, Y., Sakakura, T., Takemoto, T., Shimozono, K., Aoki, K., & Nishiyama, H. (2011). Enantioselective Addition of Allyltin Reagents to Amino Aldehydes Catalyzed with Bis(oxazolinyl)phenylrhodium(III) Aqua Complexes. Molecules, 16(7), 5387-5401. https://doi.org/10.3390/molecules16075387