An Expeditious Iodine-Catalyzed Synthesis of 3-Pyrrole-substituted 2-Azetidinones

Abstract
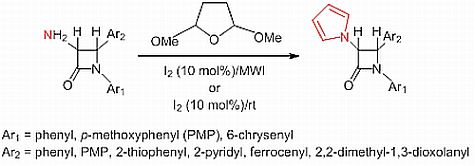
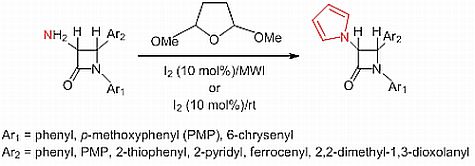
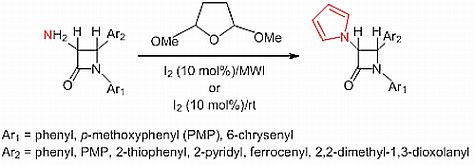
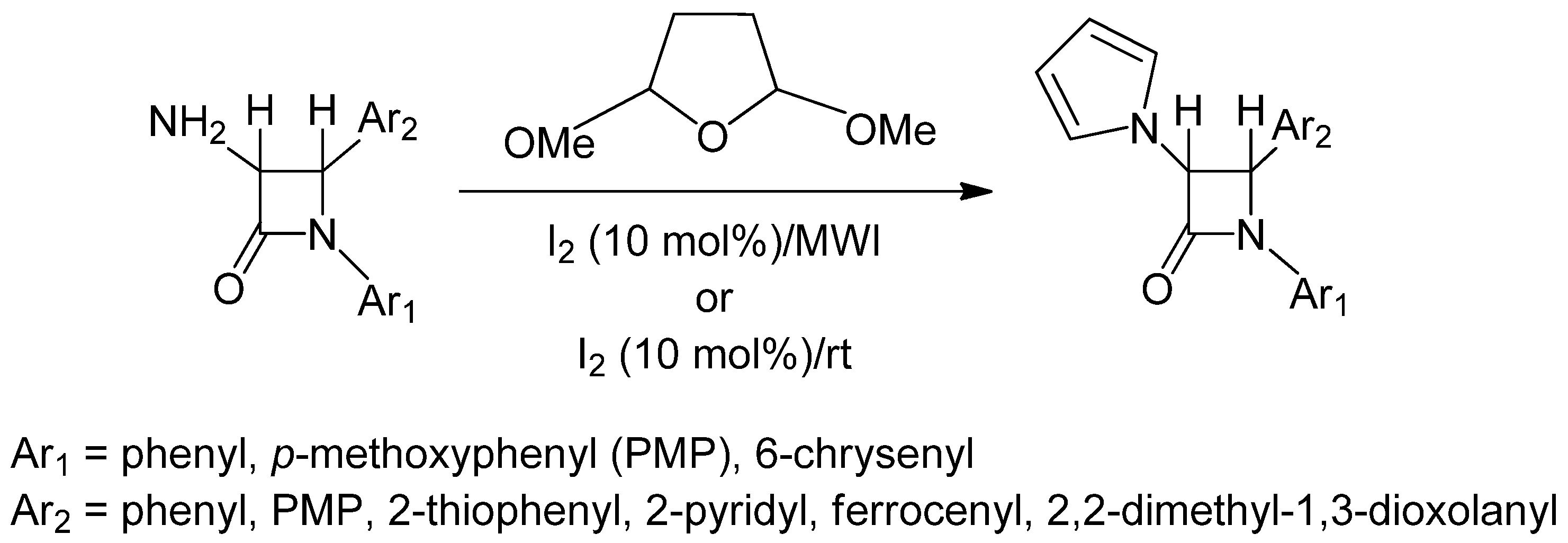
:
1. Introduction

2. Results and Discussion
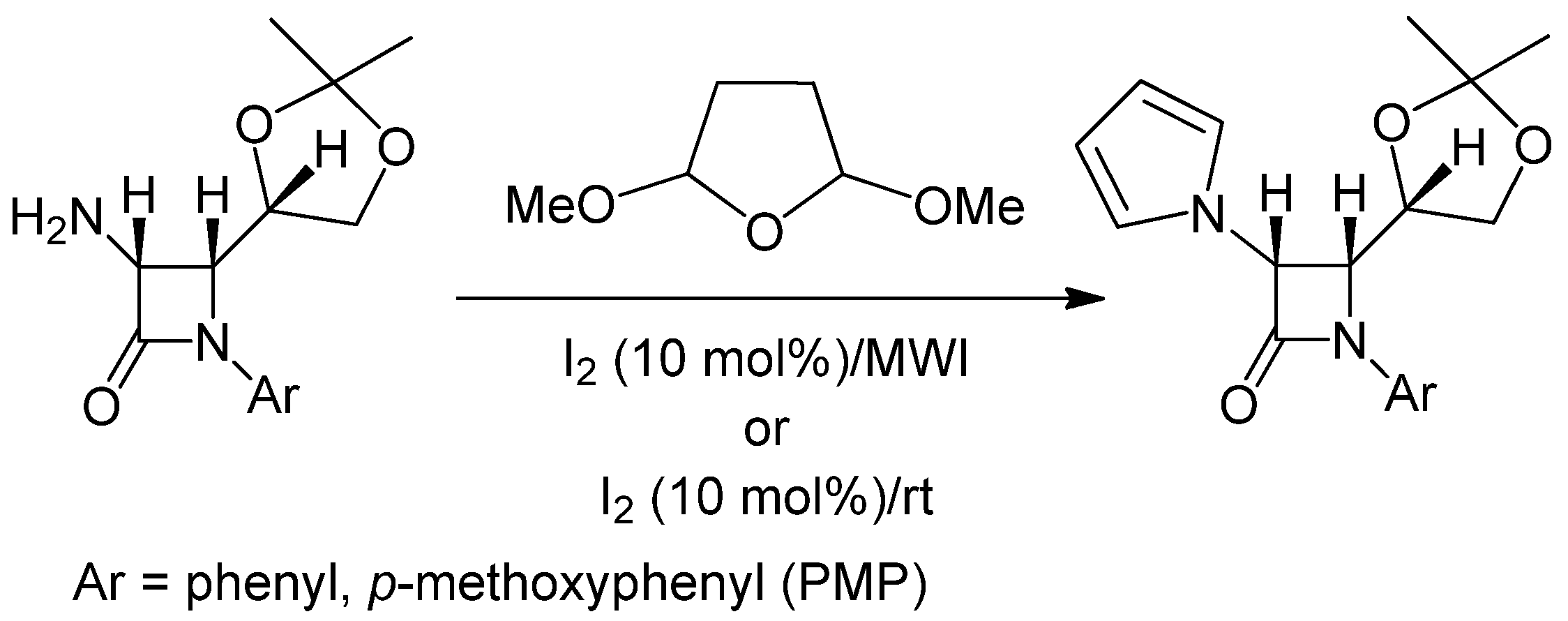
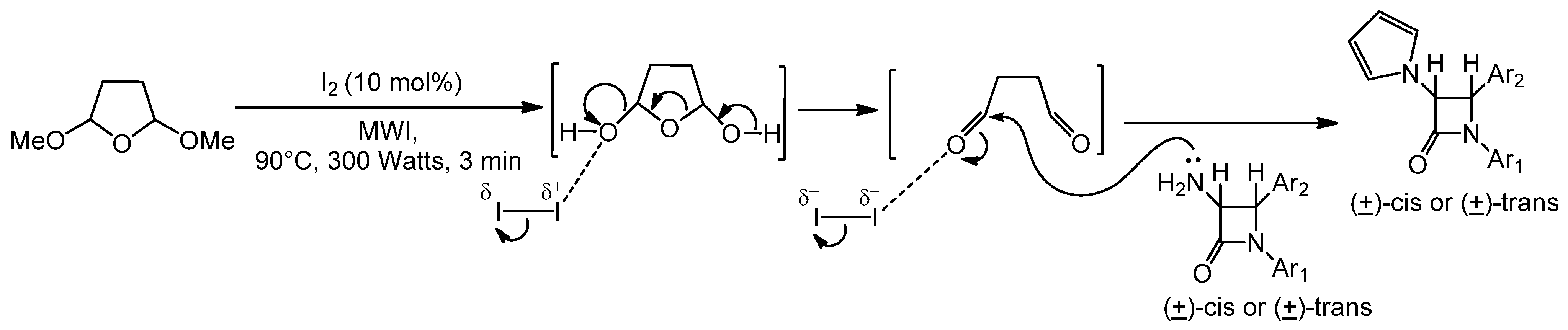
2.1. Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent (1 mL) | Yield (%) a |
|---|---|---|
| 1 | Water | 83 |
| 2 | THF | 77 |
| 3 | Ethanol | 70 |
| 4 | Toluene | 51 |
| 5 | Methanol | 69 |
| 6 | Dichloromethane | 48 |
| 7 | DMSO | 66 |
| 8 | Neat | 91 |
| Entry | Molecular I2 (mol%) | Yield (%) a |
|---|---|---|
| 1 | 30 | 78 |
| 2 | 25 | 82 |
| 3 | 20 | 91 |
| 4 | 15 | 92 |
| 5 | 10 | 98 |
| 6 | 5 | 69 |
| 7 | 2 | 54 |
| 8 | 1 | 47 |

| Entry | Substrate | Product | Condition A (MWI) | Condition B | ||
|---|---|---|---|---|---|---|
| 300 Watts/90 °C/24–50 psi | Stirring at room | |||||
| temperature | ||||||
| Time (min) | Yield (%) a | Time (h) | Yield (%) a | |||
| 1 | 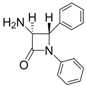 | 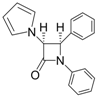 | 1 | 96 | 12 | 72 |
| 2 | 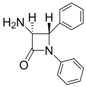 | 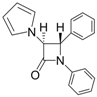 | 1 | 93 | 12 | 74 |
| 3 | 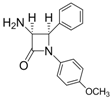 | 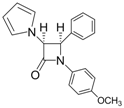 | 1 | 99 | 12 | 75 |
| 4 | 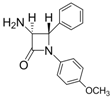 | 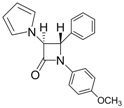 | 1 | 94 | 12 | 73 |
| 5 | 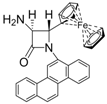 | 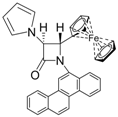 | 3 | 87 | 24 | 63 |
| 6 | 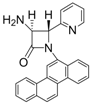 |  | 3 | 90 | 24 | 59 |
| 7 | 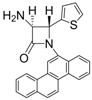 | 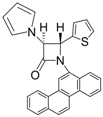 | 3 | 92 | 24 | 61 |
| 8 | 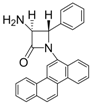 | 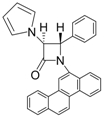 | 3 | 98 | 24 | 71 |
| 9 |  |  | 3 | 95 | 24 | 67 |
| 10 | 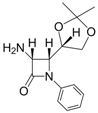 | 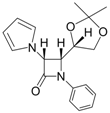 | 1 | 93 | 12 | 70 |
| 11 |  | 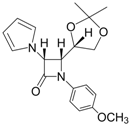 | 1 | 97 | 12 | 73 |
2.2. Discussion

3. Experimental
3.1. General
3.2. General Procedure for the Synthesis of 3-Pyrrole Substituted 2-Azetidinones from 3-Amino-2-azetidinones under Microwave Irradiation
4. Conclusions
Acknowledgments
References
- Banik, B.K.; Banik, I.; Becker, F.F. Novel anticancer β-lactams. Top. Heterocycl. Chem. 2010, 22, 349–373. [Google Scholar] [CrossRef]
- Banik, B.K.; Becker, F.F. Synthesis, electrophilic substitution and structure-activity relationship studies of polycyclic aromatic compounds towards the development of anticancer agents. Curr. Med. Chem. 2001, 8, 1513–1533. [Google Scholar]
- Bose, A.K.; Manhas, M.S.; Banik, B.K.; Srirajan, V. β-Lactams: Cyclic Amides of Distinction In The Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry, and Material Science; Greenberg, A., Breneman, C.M., Liebman, J.F., Eds.; Wiley-Interscience: New York, NY, USA, 2000; pp. 157–214, Chapter 7. [Google Scholar]
- Georg, G.I.; Ravikumar, V.T. The Organic Chemistry of β-Lactams; Georg, G.I., Ed.; VCH: New York, NY, USA, 1992. [Google Scholar]
- Alcaide, B.; Almendros, P. Allenyl-β-Lactams. Versatile scaffolds for the synthesis of heterocycles. Chem. Rec. 2011, 11, 311–330. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Aragoncillo, C. β-Lactams: versatile building blocks for the stereoselective synthesis of non-β-lactam products. Chem. Rev. 2007, 107, 4437–4492. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P. β-Lactams as versatile synthetic intermediates for the preparation of heterocycles of biological interest. Curr. Med. Chem. 2004, 11, 1921–1949. [Google Scholar]
- Deshmukh, A.R.A.S.; Bhawal, B.M.; Krishnaswamy, D.; Govande, V.V.; Shinkre, B.A.; Jayanthi, A. Azetidin-2-ones, synthon for biologically important compounds. Curr. Med. Chem. 2004, 11, 1889–1920. [Google Scholar]
- Alcaide, B.; Almendros, P. Selective bond cleavage of the β-lactam nucleus: Application in stereocontrolled synthesis. Synlett 2002, 3, 381–393. [Google Scholar] [CrossRef]
- Palomo, C.; Aizpurua, J.M.; Ganboa, I.; Oiarbide, M. β-Lactams as versatile intermediates in α- and β-amino acid synthesis. Synlett 2001, 12, 1813–1826. [Google Scholar]
- Alcaide, B.; Almendros, P. 4-Oxoazetidine-2-carbaldehydes as useful building blocks in stereocontrolled synthesis. Chem. Soc. Rev. 2001, 30, 226–240. [Google Scholar]
- Alcaide, B.; Almendros, P.; Luna, A. The chemistry of 2-azetidinones (β-Lactams). In Modern Heterocyclic Chemistry; Alvarez-Builla, J., Vaquero, J.J., Barluenga, J., Eds.; WILEY-VCH: Weinheim, Germany, 2011; Volume 4, pp. 2117–2173, Chapter 24. [Google Scholar]
- Alcaide, B.; Almendros, P.; María, C.R. Domino metal-free allene-β-lactam-based access to functionalized pyrroles. Chem. Commun. 2006, 2616–2618. [Google Scholar]
- Alcaide, B.; Almendros, P.; Carrascosa, R.; Redondo, M.C. New regiocontrolled synthesis of functionalized pyrroles from 2-azetidinone-tethered allenols. Chem. Eur. J. 2008, 14, 637–643. [Google Scholar] [CrossRef]
- Mehta, P.D.; Sengar, N.P.S.; Pathak, A.K. 2-Azetidinone—A new profile of various pharmacological activities. Eur. J. Med. Chem. 2010, 45, 5541–5560. [Google Scholar] [CrossRef]
- Staudinger, H. Ketenes. 1. Diphenylketene. Liebigs Ann. 1907, 356, 51–123. [Google Scholar] [CrossRef]
- Gupton, J.T. Pyrrole natural products with antitumor properties. Top. Heterocycl. Chem. 2006, 2, 53–92. [Google Scholar] [CrossRef]
- Arumugam, N.; Raghunathan, R.; Almansour, A.I.; Karama, U. An efficient synthesis of highly functionalized novel chromeno[4,3-b]pyrroles and indolizino[6,7-b]indoles as potent antimicrobial and antioxidant agents. Bioorg. Med. Chem. Lett. 2012, 22, 1375–1379. [Google Scholar] [CrossRef]
- Manvar, A.; Bavishi, A.; Radadiya, A.; Patel, J.; Vora, V.; Dodia, N.; Rawal, K.; Shah, A. Diversity oriented design of various hydrazides and their in vitro evaluation against Mycobacterium tuberculosis H37Rv strains. Bioorg. Med. Chem. Lett. 2011, 21, 4728–4731. [Google Scholar]
- Jiang, S.; Tala, S.R.; Lu, H.; Zou, P.; Avan, I.; Ibrahim, T.S.; Abo-Dya, N.E.; Abdelmajeid, A.; Debnath, A.K.; Katritzky, A.R. Design, synthesis, and biological activity of a novel series of 2,5-disubstituted furans/pyrroles as HIV-1 fusion inhibitors targeting gp41. Bioorg. Med. Chem. Lett. 2011, 21, 6895–6898. [Google Scholar]
- Fang, Z.; Liao, P.-C.; Yang, Y.-L.; Yang, F.-L.; Chen, Y.-L.; Lam, Y.; Hua, K.-F.; Wu, S.-H. Synthesis and biological evaluation of polyenylpyrrole derivatives as anticancer agents acting through caspases-dependent apoptosis. J. Med. Chem. 2010, 53, 7967–7978. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Mukherjee, S.; Granados, J.C.; Short, J.D.; Banik, B.K. Ultrasound-assisted bismuth nitrate-induced green synthesis of novel pyrrole derivatives and their biological evaluation as anticancer agents. Eur. J. Med. Chem. 2012, 50, 209–215. [Google Scholar] [CrossRef]
- Rudolph, D.A.; Dvorak, C.A.; Dvorak, L.; Nepomuceno, D.; Bonaventure, P.; Lovenberg, T.W.; Carruthers, N.I. Novel tetrahydropyrido[3,2-c]pyrroles as 5-HT7 antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 42–44. [Google Scholar]
- Ontoria, J.M.; Martin, H.J.I.; Malancona, S.; Attenni, B.; Stansfield, I.; Conte, I.; Ercolani, C.; Habermann, J.; Ponzi, S.; Di Filippo, M.; et al. Identification of thieno[3,2-b]pyrroles as allosteric inhibitors of hepatitis C virus NS5B polymerase. Bioorg. Med. Chem. Lett. 2006, 16, 4026–4030. [Google Scholar]
- Di Santo, R.; Tafi, A.; Costi, R.; Botta, M.; Artico, M.; Corelli, F.; Forte, M.; Caporuscio, F.; Angiolella, L.; Palamara, A.T. Antifungal agents. 11. N-Substituted derivatives of 1-[(aryl)(4-aryl-1H-pyrrol-3-yl)methyl]-1H-imidazole: Synthesis, anti-Candida activity, and QSAR studies. J. Med. Chem. 2005, 48, 5140–5153. [Google Scholar]
- Geronikaki, A.A.; Dearden, J.C.; Filimonov, D.; Galaeva, I.; Garibova, T.L.; Gloriozova, T.; Krajneva, V.; Lagunin, A.; Macaev, F.Z.; Molodavkin, G.; et al. Design of new cognition enhancers: from computer prediction to synthesis and biological evaluation. J. Med. Chem. 2004, 47, 2870–2876. [Google Scholar] [CrossRef]
- Fuerstner, A. Venturing into catalysis based natural product synthesis. Synlett 1999, 1523–1533. [Google Scholar] [CrossRef]
- Higgins, S.J. Conjugated polymers incorporating pendent functional groups-synthesis and characterization. Chem. Soc. Rev. 1997, 26, 247–258. [Google Scholar]
- McCullough, R.D.; Ewbank, P.C. Handbook of Conducting Polymers, 2nd ed.; Skotheim, T.A., Elsenbaumer, R.L., Reynolds, J.R., Dekker, M., Eds.; CRC Press: New York, NY, USA, 1998; Chapter 9. [Google Scholar]
- Becker, F.F.; Banik, B.K. Polycyclic aromatic compounds as anticancer agents: Synthesis and biological evaluation of some chrysene derivatives. Bioorg. Med. Chem. Lett. 1998, 8, 2877–2880. [Google Scholar] [CrossRef]
- Becker, F.F.; Mukhopadhyay, C.; Hackfeld, L.; Banik, I.; Banik, B.K. Polycyclic aromatic compounds as anticancer agents: Synthesis and biological evaluation of dibenzofluorene derivatives. Bioorg. Med. Chem. 2000, 8, 2693–2699. [Google Scholar] [CrossRef]
- Banik, B.K.; Becker, F.F. Polycyclic aromatic compounds as anticancer agents. 4. Structure-activity relationships of chrysene and pyrene derivatives. Bioorg. Med. Chem. 2001, 9, 593–605. [Google Scholar]
- Bandyopadhyay, D.; Granados, J.C.; Short, J.D.; Banik, B.K. Polycyclic aromatic compounds as anticancer agents: Evaluation of synthesis and in vitro cytotoxicity. Oncol. Lett. 2012, 3, 45–49. [Google Scholar]
- Banik, B.K.; Becker, F.F.; Banik, I. Synthesis of anticancer β-lactams: Mechanism of action. Bioorg. Med. Chem. 2004, 12, 2523–2528. [Google Scholar] [CrossRef]
- Banik, I.; Becker, F.F.; Banik, B.K. Stereoselective synthesis of β-lactams with polyaromatic imines: Entry to new and novel anticancer agents. J. Med. Chem. 2003, 46, 12–15. [Google Scholar] [CrossRef]
- Banik, B.K.; Banik, I.; Becker, F.F. Stereocontrolled synthesis of anticancer β-lactams via the Staudinger reaction. Bioorg. Med. Chem. 2005, 13, 3611–3622. [Google Scholar] [CrossRef]
- Tekale, S.U.; Kauthale, S.S.; Dake, S.A.; Sarda, S.R.; Pawar, R.P. Molecular iodine, an efficient and versatile reagent for organic synthesis. Curr. Org. Chem. 2012, 16, 1485–1501. [Google Scholar] [CrossRef]
- Parvatkar, P.T.; Parameswaran, P.S.; Tilve, S.G. Recent developments in the synthesis of five- and six-membered heterocycles using molecular iodine. Chem. Eur. J. 2012, 18, 5460–5489. [Google Scholar] [CrossRef]
- Veisi, H. Molecular iodine: Recent application in heterocyclic synthesis. Curr. Org. Chem. 2011, 15, 2438–2468. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Organoiodine (V) reagents in organic synthesis. J. Org. Chem. 2011, 76, 1185–1197. [Google Scholar] [CrossRef]
- Togo, H.; Iida, S. Synthetic use of molecular iodine for organic synthesis. Synlett 2006, 14, 2159–2175. [Google Scholar] [CrossRef]
- Das, S.; Borah, R.; Devi, R.R.; Thakur, A.J. Molecular iodine in protection and deprotection chemistry. Synlett 2008, 18, 2741–2762. [Google Scholar]
- Alcaide, B.; Almendros, P.; Cabrero, G.; Callejo, R.; Ruiz, M.P.; Arno, M.; Domingo, L.R. Ring expansion versus cyclization in 4-oxoazetidine-2-carbaldehydes catalyzed by molecular iodine: experimental and theoretical study in concert. Adv. Synth. Catal. 2010, 352, 1688–1700. [Google Scholar] [CrossRef]
- Chen, C.-C.; Yang, S.-C.; Wu, M.-J. Iodine-mediated cascade cyclization of enediynes to iodinated benzo[a]carbazoles. J. Org. Chem. 2011, 76, 10269–10274. [Google Scholar] [CrossRef]
- Lee, W.-C.; Shen, H.-C.; Hu, W.-P.; Lo, W.-S.; Murali, C.; Vandavasi, J.K.; Wang, J.-J. Iodine-catalyzed, stereo- and regioselective synthesis of 4-arylidine-4H-benzo[d][1,3]oxazines and their applications for the synthesis of quinazoline 3-oxides. Adv. Synth. Catal. 2012, 354, 2218–2228. [Google Scholar] [CrossRef]
- Banik, B.K.; Zegrocka, O.; Manhas, M.S.; Bose, A.K. Studies on lactams. 104. Enantiomerically pure β-lactams with the thienamycin side chain via glycosylation. Heterocycles 1997, 27, 173–176. [Google Scholar]
- Banik, B.K.; Manhas, M.S.; Bose, A.K. Studies on lactams. 103. Enantiopure α-hydroxy-β-lactams via stereoselective glycosylation. Tetrahedron Lett. 1997, 38, 5077–5080. [Google Scholar] [CrossRef]
- Banik, B.K.; Fernandez, M.; Alvarez, C. Iodine-catalyzed highly efficient Michael reaction of indoles under solvent-free condition. Tetrahedron Lett. 2005, 46, 2479–2482. [Google Scholar] [CrossRef]
- Banik, B.K.; Garza, R. Molecular iodine-catalyzed deprotection of acetals and ketals in acetone. Chem. Edu. 2007, 12, 75–76. [Google Scholar]
- Bandyopadhyay, D.; Mukherjee, S.; Banik, B.K. An expeditious synthesis of N-substituted pyrroles via microwave-induced iodine-catalyzed reaction under solventless conditions. Molecules 2010, 15, 2520–2525. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Mukherjee, S.; Rodriguez, R.R.; Banik, B.K. An effective microwave-induced iodine-catalyzed method for the synthesis of quinoxalines via condensation of 1,2-dicarbonyl compounds. Molecules 2010, 15, 4207–4212. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Banik, B.K. Microwave-induced stereoselectivity of β-lactam formation with dihydrophenanthrenyl imines via Staudinger cycloaddition. Helv. Chim. Acta 2010, 93, 298–301. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Yañez, M.; Banik, B.K. Microwave-induced stereoselectivity of β-lactam formation: effects of solvents. Heterocycl. Lett. 2011, 1, 65–67. [Google Scholar]
- Bandyopadhyay, D.; Rivera, G.; Salinas, I.; Aguilar, H.; Banik, B.K. Iodine-catalyzed remarkable synthesis of novel N-polyaromatic β-lactams bearing pyrroles. Molecules 2010, 15, 1082–1088. [Google Scholar] [CrossRef]
- Andoh-Baidoo, R.; Danso, R.; Mukherjee, S.; Bandyopadhyay, D.; Banik, B.K. Microwave-induced N-bromosuccinimide-mediated novel synthesis of pyrroles via Paal-Knorr reaction. Heterocycl. Lett. 2011, 1, 107–109. [Google Scholar]
- Bandyopadhyay, D.; Banik, A.; Bhatta, S.; Banik, B.K. Microwave-assisted ruthenium trichloride catalyzed synthesis of pyrroles fused with indole systems. Heterocycl. Commun. 2009, 15, 121–122. [Google Scholar]
- Abrego, D.; Bandyopadhyay, D.; Banik, B.K. Microwave-induced indium-catalyzed synthesis of pyrrole fused with indolinone in water. Heterocycl. Lett. 2011, 1, 94–95. [Google Scholar]
- Rivera, S.; Bandyopadhyay, D.; Banik, B.K. Facile synthesis of N-substituted pyrroles via microwave-induced bismuth nitrate-catalyzed reaction under solventless conditions. Tetrahedron Lett. 2009, 50, 5445–5448. [Google Scholar] [CrossRef]
- Kall, A.; Bandyopadhyay, D.; Banik, B.K. Microwave-induced aza-Michael reaction in water: A remarkable simple procedure. Synth. Commun. 2010, 42, 1730–1735. [Google Scholar]
- Bandyopadhyay, D.; Fonseca, R.S.; Banik, B.K. Microwave-induced bismuth nitrate-mediated selective hydrolysis of amide. Heterocycl. Lett. 2011, 1, 75–77. [Google Scholar]
- Iglesias, L.; Aguilar, C.; Bandyopadhyay, D.; Banik, B.K. A new bismuth nitrate-catalyzed electrophilic substitution of indoles with carbonyl compounds under solventless conditions. Synth. Commun. 2010, 40, 3678–3682. [Google Scholar] [CrossRef]
- Rivera, S.; Bandyopadhyay, D.; Banik, B.K. Microwave-induced bismuth nitrate-catalyzed electrophilic substitution of 7-aza indole with activated carbonyl compound under solvent-free conditions. Heterocycl. Lett. 2011, 1, 43–46. [Google Scholar]
- Rivera, G.; Bandyopadhyay, D.; Jaggi, S.; Gonzales, R.C.; Banik, B.K. An expeditious synthesis of 3-amino β-lactams derived from polyaromatic compounds. Heterocycl. Commun. 2009, 15, 323–325. [Google Scholar]
- Bandyopadhyay, D.; Xavier, M.; Banik, B.K. Highly stereoselective β-lactam synthesis via the Staudinger reaction using polyaromatic imines. Heterocycl. Commun. 2009, 15, 229–232. [Google Scholar]
- Manhas, M.S.; Amin, S.G.; Ram, B.; Bose, A.K. Studies on lactams. Part L. Annulation of imines to β-lactams at low temperatures. Synthesis 1976, 10, 689–690. [Google Scholar]
- Bose, A.K.; Chiang, Y.H.; Manhas, M.S. β-Lactams. XXI. Mechanism of β-lactam formation. Tetrahedron Lett. 1972, 40, 4091–4094. [Google Scholar]
- Miyake, M.; Kirisawa, M.; Tokutake, N. A convenient synthesis of β-lactams. Synthesis 1982, 12, 1053–1056. [Google Scholar]
- Martins, M.A.P.; Frizzo, C.P.; Moreira, D.N.; Buriol, L.; Machado, P. Solvent-free heterocyclic synthesis. Chem. Rev. 2009, 109, 4140–4182. [Google Scholar]
- Kidwai, M. Green chemistry trends toward sustainability. Pure Appl. Chem. 2006, 78, 1983–1992. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Maldonado, S.; Banik, B.K. A microwave-assisted bismuth nitrate-catalyzed unique route toward 1,4-dihydropyridines. Molecules 2012, 17, 2643–2662. [Google Scholar] [CrossRef]
- Kranjc, K.; Kočevar, M. Microwave-assisted organic synthesis: General considerations and transformations of heterocyclic compounds. Curr. Org. Chem. 2010, 14, 1050–1074. [Google Scholar] [CrossRef]
- Blackwell, H.E. Out of the oil bath and into the oven—Microwave-assisted combinatorial chemistry heats up. Org. Biomol. Chem. 2003, 1, 1251–1255. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Cruz, J.; Banik, B.K. Novel synthesis of 3-pyrrole substituted β-lactams via microwave-induced bismuth nitrate-catalyzed reaction. Tetrahedron 2012. Ahead of Print. [Google Scholar]
- Sample Availability: Samples of the all compounds (mg quantity) are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bandyopadhyay, D.; Cruz, J.; Yadav, R.N.; Banik, B.K. An Expeditious Iodine-Catalyzed Synthesis of 3-Pyrrole-substituted 2-Azetidinones. Molecules 2012, 17, 11570-11584. https://doi.org/10.3390/molecules171011570
Bandyopadhyay D, Cruz J, Yadav RN, Banik BK. An Expeditious Iodine-Catalyzed Synthesis of 3-Pyrrole-substituted 2-Azetidinones. Molecules. 2012; 17(10):11570-11584. https://doi.org/10.3390/molecules171011570
Chicago/Turabian StyleBandyopadhyay, Debasish, Jessica Cruz, Ram N. Yadav, and Bimal K. Banik. 2012. "An Expeditious Iodine-Catalyzed Synthesis of 3-Pyrrole-substituted 2-Azetidinones" Molecules 17, no. 10: 11570-11584. https://doi.org/10.3390/molecules171011570
APA StyleBandyopadhyay, D., Cruz, J., Yadav, R. N., & Banik, B. K. (2012). An Expeditious Iodine-Catalyzed Synthesis of 3-Pyrrole-substituted 2-Azetidinones. Molecules, 17(10), 11570-11584. https://doi.org/10.3390/molecules171011570