Multi-Component One-Pot Synthesis and Antimicrobial Activities of 3-Methyl-1,4-diphenyl-7-thioxo-4,6,8,9-tetrahydro-pyrazolo[5,4-b]pyrimidino[5,4-e]pyridine-5-one and Related Derivatives

Abstract
:1. Introduction
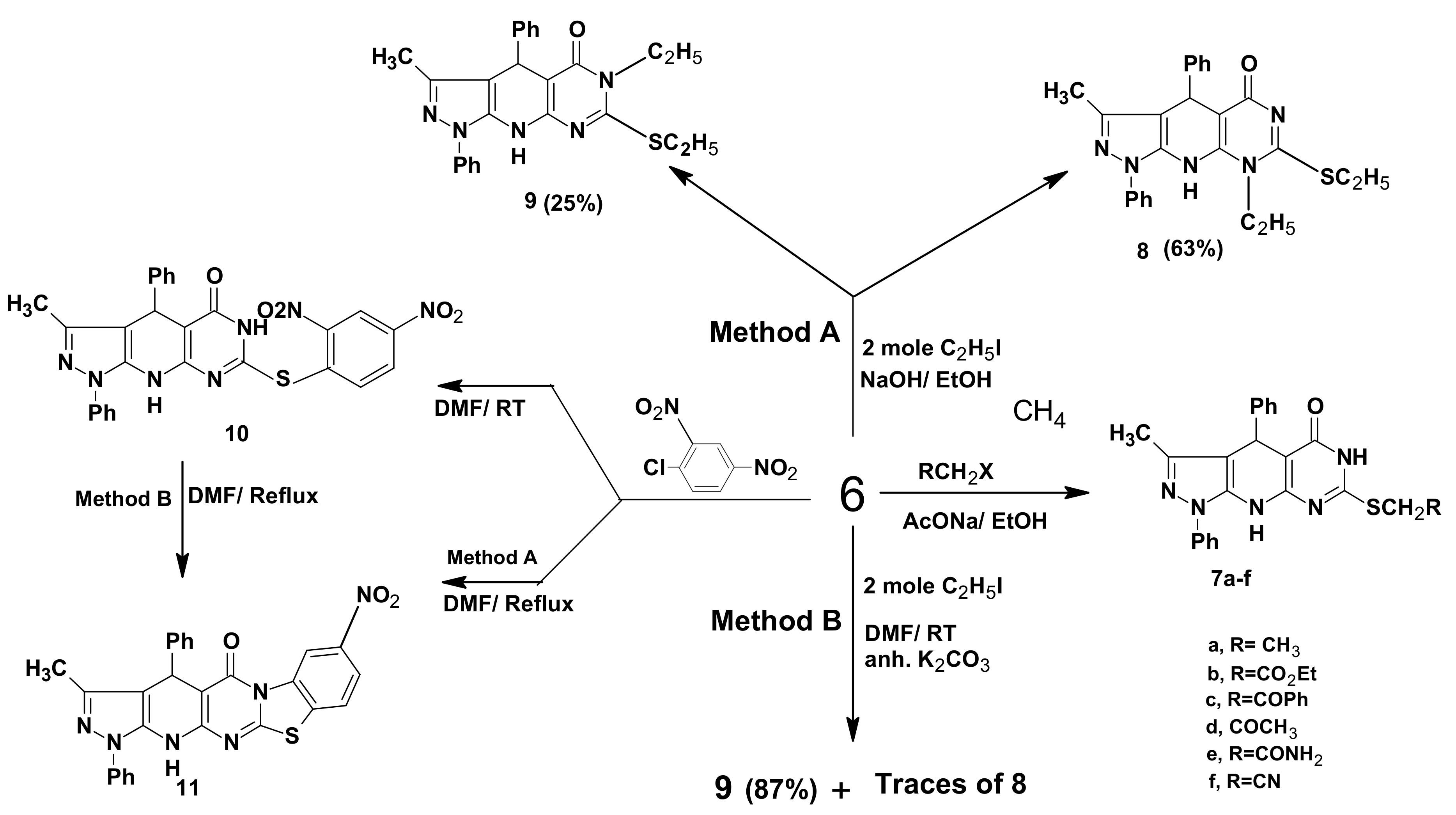
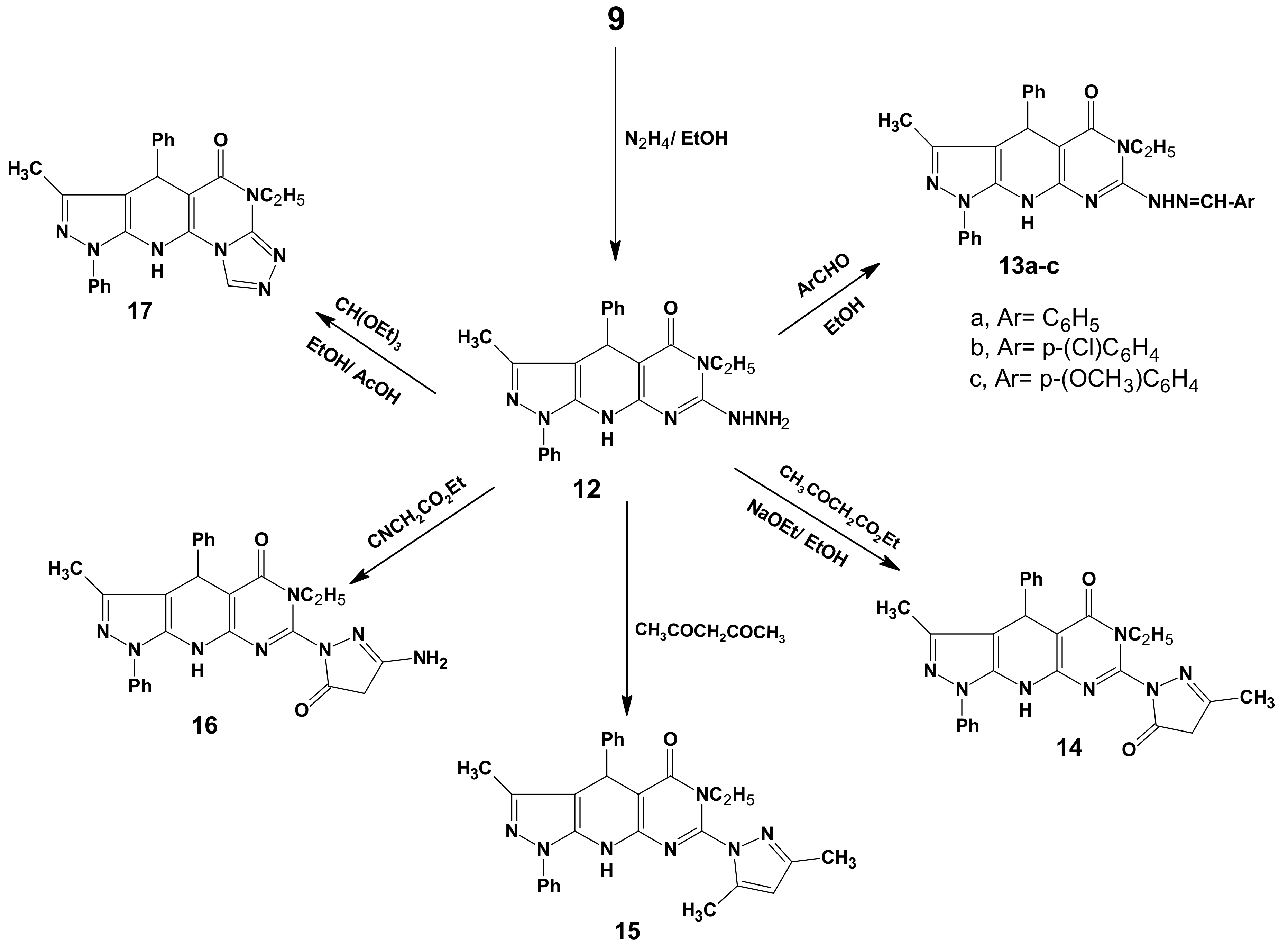
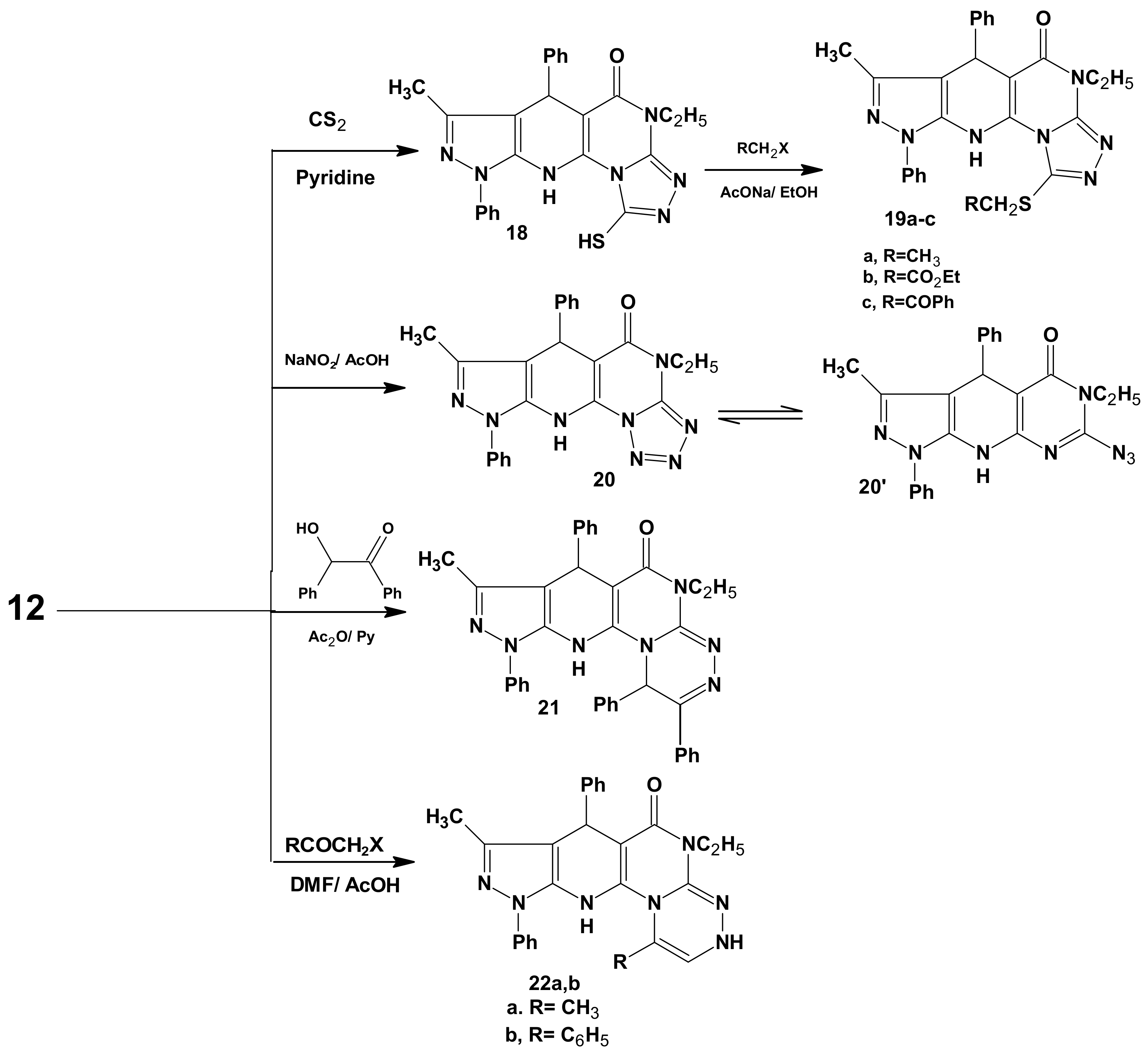
2. Results and Discussion
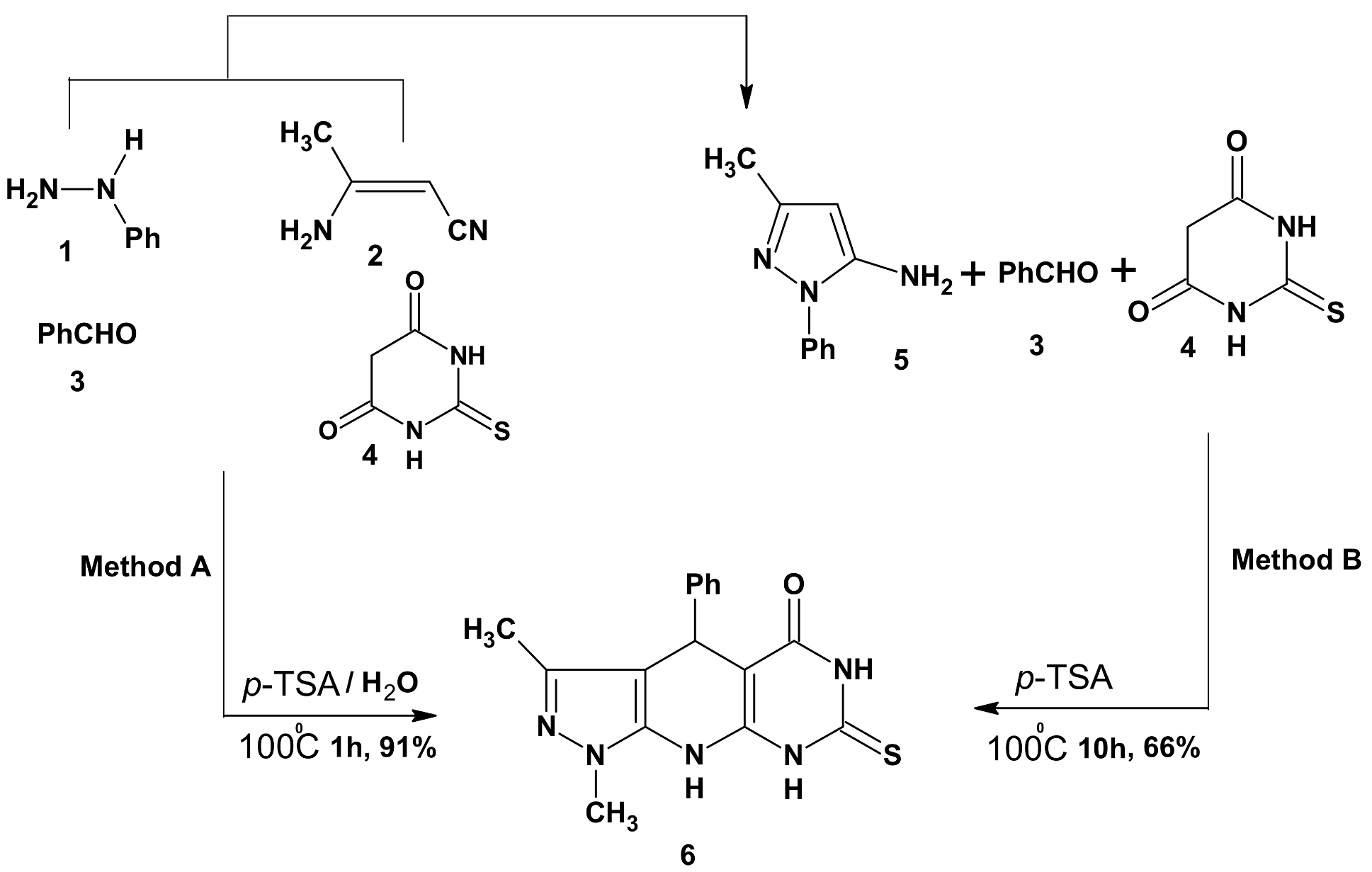
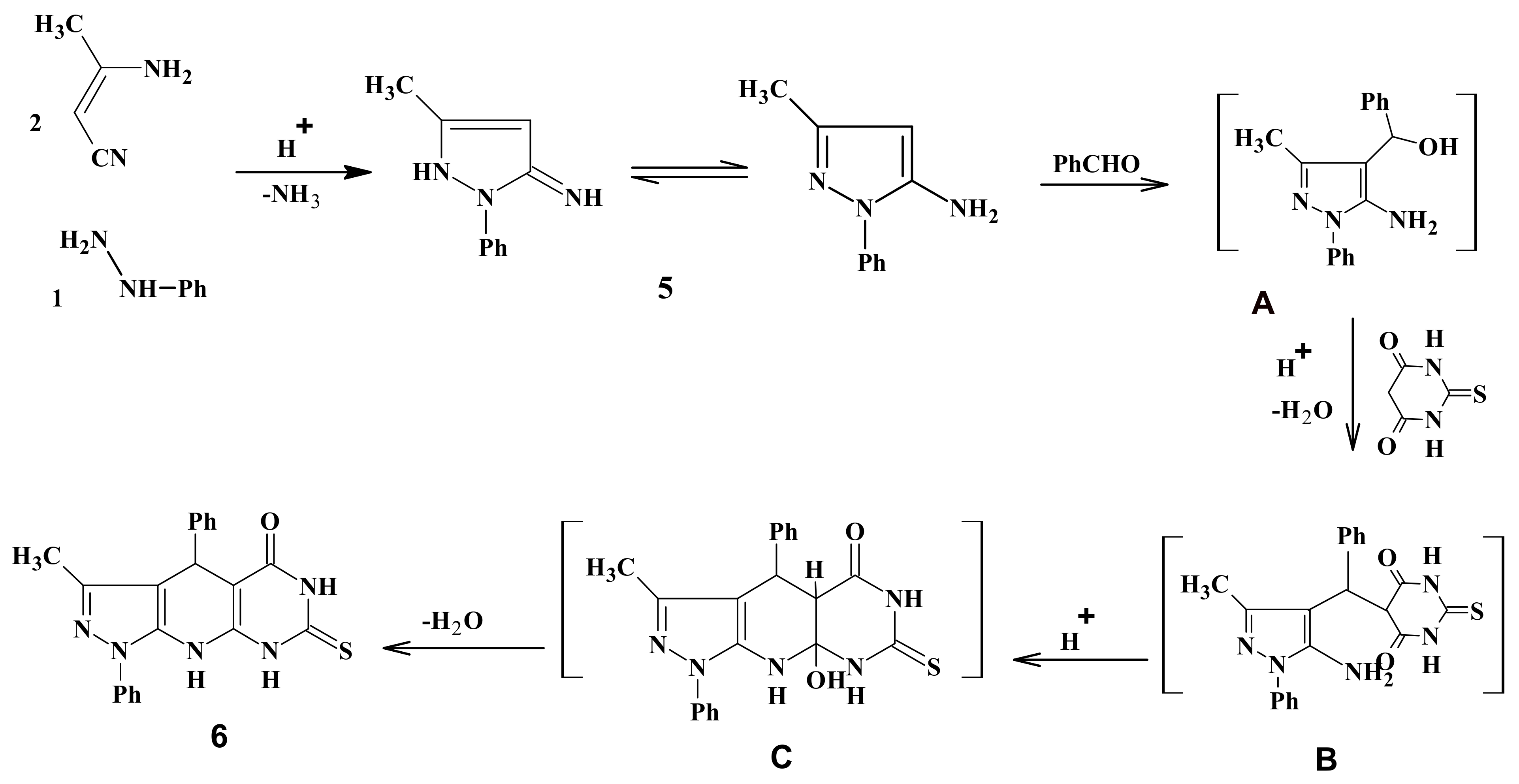
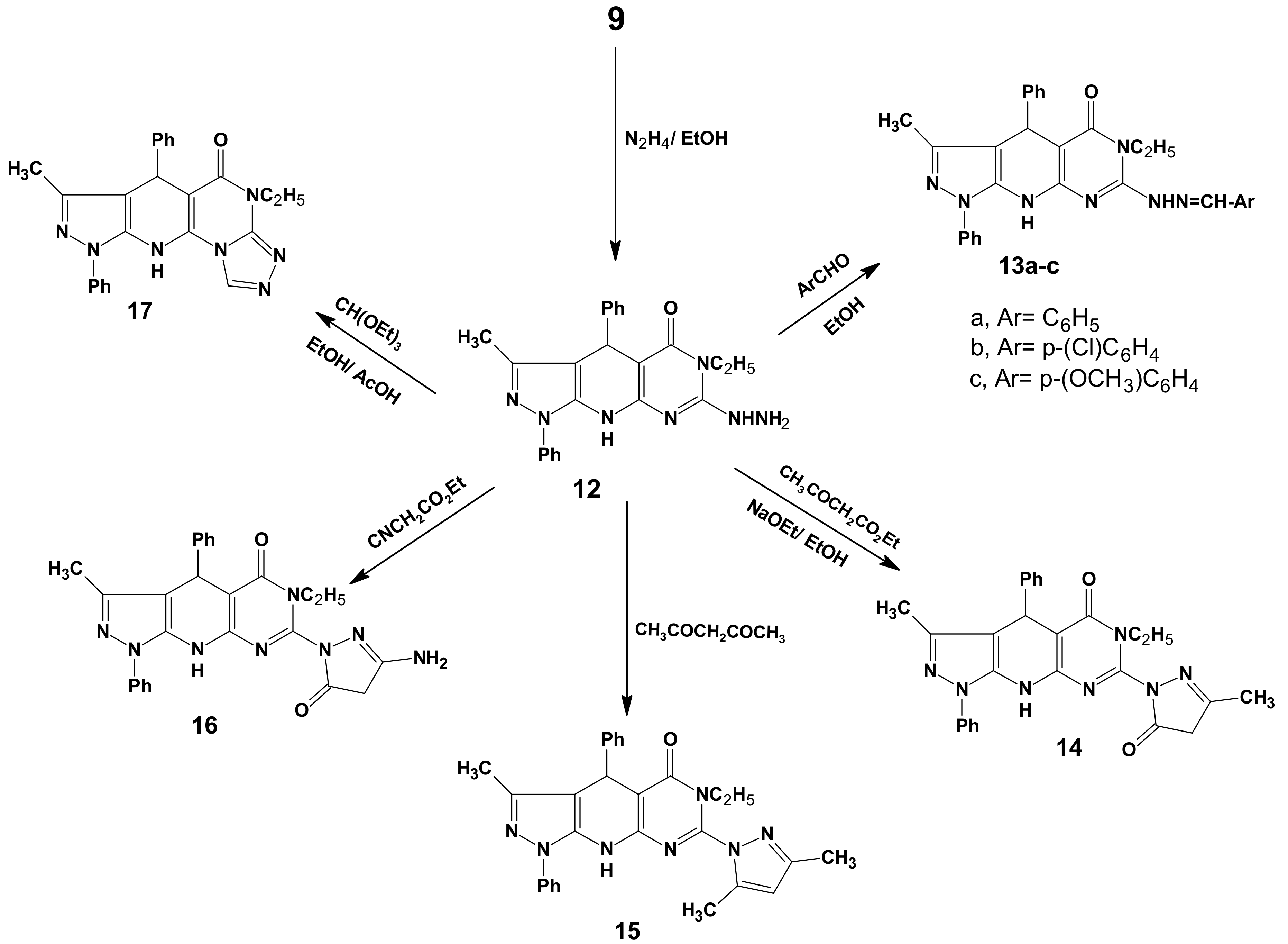
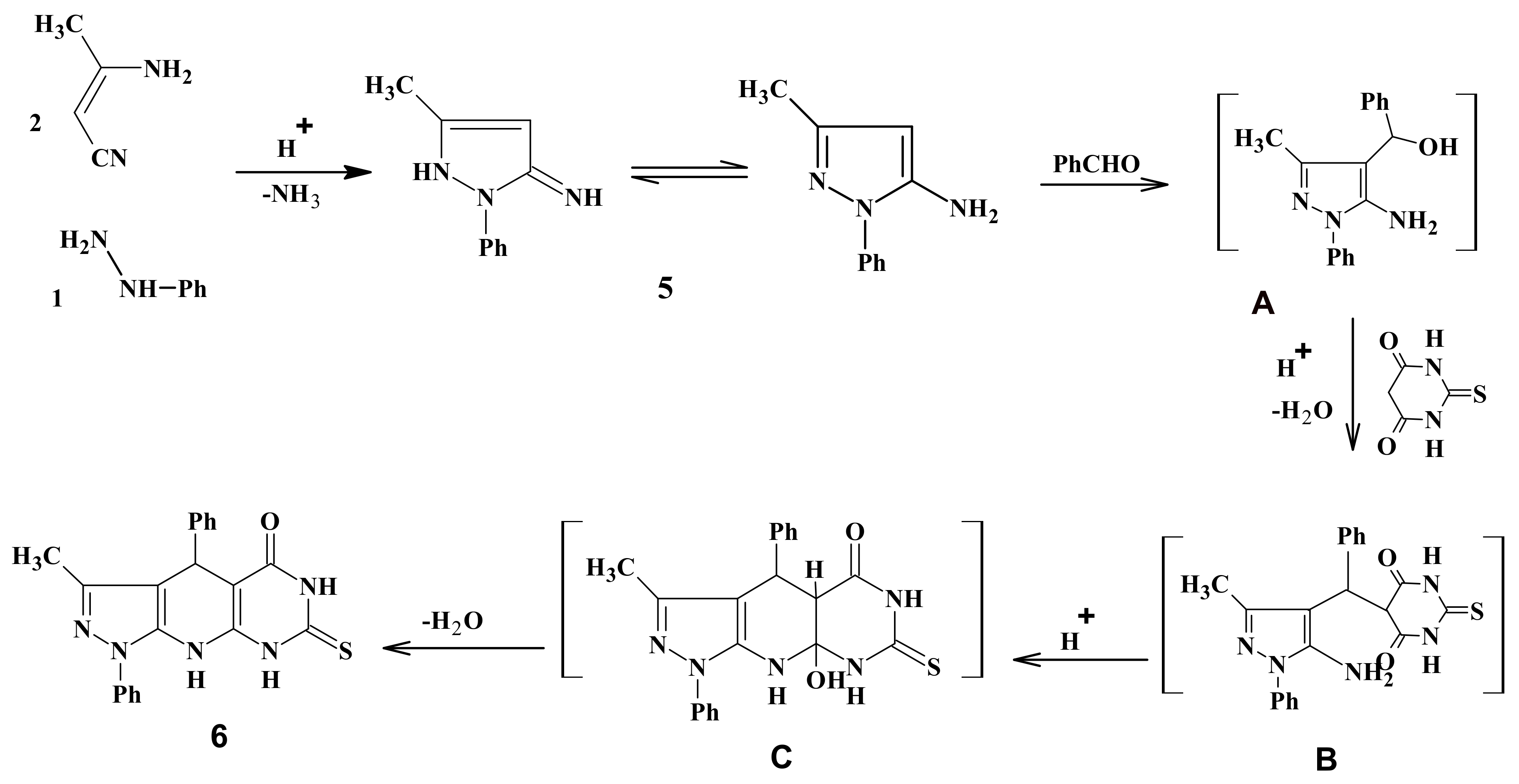
2.1. Chemistry
2.2. Biological Results
3. Experimental
3.1. General
3.2. Synthesis of 3-Methyl-1,4-diphenyl-7-thioxo-4,6,8,9-tetrahydropyrazolo[5,4-b]pyrimidino[5,4-e]pyridin-5-one (6)
3.3. General Procedure for the Preparation of 7a–f
3.4. 8-Ethyl-7-ethylthio-3-methyl-1,4-diphenyl-4,6,9-trihydropyrazolo[5,4-b]pyrimidino[5,4-e]pyridin-5-one (8) and 6-Ethyl-7-ethylthio-3-methyl-1,4-diphenyl-4,6,9-trihydropyrazolo[5,4-b]pyrimidino[5,4-e]pyridin-5-one (9)
3.5. 7-(2,4-Dinitrophenylthio)-3-methyl-1,4-diphenyl-4,6,9-trihydropyrazolo[5,4-b]pyrimidino[5,4-e] pyridin-5-one (10)
3.6. 3-Methyl-8-nitro-1,4-diphenyl-4,6,13-trihydrobenzothiazolo[3',2'-2,1]pyrimidino[5,4-e] pyrazolo-[5,4-b]pyridin-5-one (11)
3.7. 6-Ethyl-7-hydrazino-3-methyl-1,4-diphenyl-4,6,9-trihydropyrazolo[5,4-b]pyrimidino[5,4-e]-pyridin-5-one (12)
3.8. General Procedure for the Preparation of 13a–c
3.9. 6-Ethyl-3-methyl-7-(3-methyl-5-oxo(2-pyrazolinyl))-1,4-diphenyl-4,6,9-trihydropyrazolo[5,4-b]-pyrimidino[5,4-e]pyridin-5-one (14)
3.10. 7-(3,5-Dimethylpyrazolyl)-6-ethyl-3-methyl-1,4-diphenyl-4,6,9-trihydropyrazolo[5,4-b]-pyrimidine[5,4-e]pyridin-5-one (15)
3.11. 7-(3-Amino-5-oxo(2-pyrazolinyl))-6-ethyl-3-methyl-1,4-diphenyl-4,6,9-tri-hydropyrazolo[5,4-b]-pyrimidino[5,4-e]pyridin-5-one (16)
3.12. 11-Ethyl-8-methyl-6,9-diphenyl-4,5,9,11-tetrahydropyrazolo[5,4-b]1,2,4-triazolo[4',3'-2,1]-pyrimidino[5,6-e]pyridin-10-one (17)
3.13. 11-Ethyl-8-methyl-6,9-diphenyl-10-oxo-4,5,9,11-tetrahydropyrazolo[5,4-b]1,2,4-triazolo[4',3'-2,1]pyrimidino[5,6-e]pyridin-3(2H)-thione (18)
3.14. General Procedure for the Preparation of 19a–c
3.15. 4-Ethyl-7-methyl-6,9-diphenyl-4,6,10,11-tetrahydropyrazolo[5,4-b]1,2,3,4-tetraazolo[1',5'-1,2]-pyrimidino[5,6-e]pyridin-5-one (20)
3.16. 5-Ethyl-8-methyl-1,2,7,10-tetraphenyl-5,7,11,12-tetrahydro-1H-pyrazolo[5,4-b]1,2,4-triazino[4',3'-2,1]-pyrimidino[5,6-e]pyridin-6-one (21)
3.17. General Procedure for the Preparation of 22a,b
3.18. Antimicrobial Activity
4. Conclusions
Supplementary Materials
Acknowledgements
References
- Ganem, B. Strategies for innovation in multicomponent reaction design. Acc. Chem. 2009, 42, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Padwa, A. Domino reactions of rhodium (II) carbenoids for alkaloid synthesis. Chem. Soc. Rev. 2009, 38, 3072–3081. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Amir, T.M.; Morteza, R.; Aram, R. Novel three-component reaction of a secondary amine and a 2-hydroxybenzaldehyde derivative with an isocyanide in the presence of silica gel: an efficient one-pot synthesis of benzo[b]furan derivatives. Tetrahedron Lett. 2009, 50, 5625–5627. [Google Scholar]
- Zeinab, Z.; Mehdi, K.; Ali, R.; Alireza, F.; Ali, S.; Katarzyna, Ś.; Tadeusz Lis, A.S. Synthesis of functionalized furo[3,2-c]coumarins via a one-pot oxidative pseudo three-component reaction in poly(ethylene glycol. Tetrahedron 2012, 68, 6721–6726. [Google Scholar]
- Domling, A. Multicomponent reactions. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [PubMed]
- D'Souza, D.M.; Muller, T.J. Multi-component syntheses of heterocycles by transition-metal catalysis. J. Chem. Soc. Rev. 2007, 36, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Ali, S. Iminophosphorane-mediated one-pot synthesis of 1,3,4-oxadiazole Derivatives. ARKIVOC 2008, xvi, 235–242. [Google Scholar]
- Ali, S.; Ali, R.; Nouri, B.; Richard, W. The reaction of (N-isocyanimino)triphenylphosphorane with dialkyl acetylenedicarboxylates in the presence of 1,3-diphenyl-1,3-propanedione: A novel three-component reaction for the stereoselective synthesis of dialkyl (Z)-2-(5,7-diphenyl-1,3,4-oxadiazepin-2-yl)-2-butenedioates. Tetrahedron Lett. 2007, 48, 2617–2620. [Google Scholar]
- Ali, S.; Ali, R. The reaction of (N-isocyanimino)triphenylphosphorane with benzoic acid derivatives: A novel synthesis of 2-aryl-1,3,4-oxadiazole derivatives. Tetrahedron Lett. 2007, 48, 1549–1551. [Google Scholar]
- Shore, G.; Yoo, W.J.; Li, C.J.; Organ, M. Propargyl amine synthesis catalysed by gold and copper thin films using microwave assistant, continuous flow organic synthesis. Chem. Eur. J. 2010, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.R. The chemistry of pyrazolopyridine. Adv. Heterocyl. Chem. 1984, 36, 343–409. [Google Scholar]
- Elnagdi, M.H.; Elgemeie, G.H.; El-Moghayar, R.M.H. Chemistry of pyrazolopyrimidines. Adv. Heterocycl. Chem. 1987, 41, 319–376. [Google Scholar]
- Jiaro, Q.; Jaime, P.; Silva, C.; Rodrigo, A.; Braulio, I.; Manuel, N.; Justo, C.; Mike, H. Solvent free synthesis of fused pyrazolo [1,5-a]pyrimidines by reaction of 5-amino-1-H-pyrazoles and β-triketones. Open Org. Chem. J. 2008, 2, 92–99. [Google Scholar]
- Jiaro, Q.; Jaime, P.; Rodrigo, A.; Manuel, N.; Justo, C.; Mike, H. Regioselective synthesis of novel substituted pyrazolo[1,5-a]pyrimidines under solvent-free conditions. Tetrahedron Lett. 2008, 49, 6254–6256. [Google Scholar]
- Jiaro, Q.; Jaime, P.; Hugo, S.; Rodrigo, A.; Braulio, I.; Manuel, N.; Justo, C. Regioselective synthesis of fused benzopyrazolo[3,4-b]quinolines under solvent-free conditions. Tetrahedron Lett. 2007, 48, 1987–1990. [Google Scholar]
- Aly, M.F.; El-Naggar, G.M.; El-Emary, T.I.; Girgg, R.; Metwally, S.A.; Sivagnanam, S. X=Y-ZH Compounds as potential 1,3-Dipoles part 41. Azomethine Ylide Formation from the reaction of -Amino acids and esters wth Alloxan (Strecker Degradation) and with 1-phenyl-3-methyl pyrazoline 4,5-dione. Tetrahedron 1994, 50, 895–906. [Google Scholar]
- Selleri, S.; Burni, F.; Castanzo, A.; Gueririni, G.; Malmbeg-Aiello, P.; Lavarone, G.; Martini, C. Synthesis and preliminary evaluation of pyrazolo[1,5-a]pyrido[3,4-e]pyrimidin-6-(7H)-ones and related comounds as benzodiazepine receptor ligands and anticonvulsant agents. Eur. J. Med. Chem. 1992, 27, 985–990. [Google Scholar] [CrossRef]
- Poreba, K.; Wietrzyk, J.; Opolski, A. Synthesis and antiproliferative activity in vitro of new 2,3 or 4 substituted pyrido[2',3':3,4]pyrazolo[1,5-a]pyrimidines. Acta Pol. Pharm. 2006, 63, 189–194. [Google Scholar] [PubMed]
- Ismail, M.M.F.; Ammar, Y.A.; El-Zahaby, H.S.A.; Eisa, S.I.; Barakat, S.E. Synthesis of Novel-1-pyrazolylpyridinopyrimidin2-ones as potential Anti-inflammatory and Analgesic Agent. Arch. Pharm. Chem. Life Sci. 2007, 340, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Stoy, P.; He, B.; Adam, L.; Krupinski, J.; Normandin, D.; Pongrac, R.; Seliger, L.; Waston, A.; Macor, J.E. The discovery of novel, potent and selective PDE inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 2461–2464. [Google Scholar] [CrossRef]
- Ahluwalia, V.K.; Dahiya, A.; Garg, V. Reaction of 5-amino-4-formyl-3-methyl(or phenyl)-1-phenyl-1H-pyrazoles with active methylene compounds: Synthesis of fused heterocyclic rings. 1997, 36B, 88–90, and references sited therein. [Google Scholar] [CrossRef]
- Metwally, S.A.; El-Naggar, G.M.; El-Emary, T.I. Reactions of 4-(Dicyanomethylene)-3-methyl-1-phenyl-2-pyrazolin-5-one towards methylene comoponds. Liebgs Ann. Chem. 1991, 62, 961–962. [Google Scholar] [CrossRef]
- Metwally, S.A.; El-Naggar, G.M.; Younis, M.I.; Elnagdi, M.H.; El-Emary, T.I. Reactions of 4-(Dicyanomethylene)-3-methyl-1-phenyl-2-pyrazolin-5- one towards Amines and Phenols. Liebgs Ann. Chem. 1989, 40, 1037–1040. [Google Scholar] [CrossRef]
- El-Emary, T.I.; El-Dean, A.M.; El-Kashef, H.S. Facile synthesis of some new pyrazolo [3,4-b] pyrazines and their antifungal activity. II Farmaco 1998, 53, 383–388. [Google Scholar] [CrossRef]
- El-Kashef, H.S.; El-Emary, T.I.; Gasquet, M.; Timon-David, M.J.; Vanele, P. New pyrazolo [3,4-b] pyrazines: Synthesis and biological activity. Pharmazie 2000, 55, 572–577. [Google Scholar] [PubMed]
- El-Emary, T.I.; El-kashef, H.S.; Hussein, A.M. New Polycyclic Azines Derived from Pyrazolo [3,4-b] pyridine. Pharmazie 2000, 55, 356–358. [Google Scholar] [PubMed]
- Hussein, A.M.; El-Emary, T.I. Polycyclic Pyrazoles: Routes to New Pyrazoloazines. J. Chem. Res. 1998, 228–236. [Google Scholar] [CrossRef]
- El-Emary, T.I.; Bakhite, E.A. Synthesis and biological screening of new 1,3- diphenylpyrazoles with different moieties at position-4. Pharmazie 1999, 2, 106–111. [Google Scholar]
- El-Emary, T.I.; Abdel-Mohsen, Sh.A. Synthesis and antimicrobial activity of some new 1,3-diphenyl pyrazoles bearing pyrimidine, pyrimidinethione, thiazolopyrimidine, triazolopyrimidine, thio and alkyl-thiotriazolpyrimidinone moieties at 4- position. Phosphorous Sulfur Silicon 2006, 181, 2459–2474. [Google Scholar] [CrossRef]
- El-Emary, T.I.; Khalil, A.; Ali, G.A.; El-Adasy, A.A. A facile synthesis of some new Thiazolo[3,2-a]pyridines containing pyrazolyl moiety and their antimicrobial activity. Phosphorous Sulfur Silicon 2005, 180, 19–30. [Google Scholar] [CrossRef]
- El-Emary, T.I. Synthesis of newly substituted pyrazoles and substituted pyrazolo[3,4-b]pyridines based on 5-amino-3-methyl-1-phenylpyrazole. J. Chin. Chem. Soc. 2007, 54, 507–518. [Google Scholar] [CrossRef]
- El-Emary, T.I. Synthesis, reactions and biological activity of some new pyrazolo [3,4-b] pyrazines. J. Chin. Chem. Soc. 2006, 53, 391–401. [Google Scholar] [CrossRef]
- El-Emary, T.I. Synthesis of some newly condensed and uncondensed pyrazolo[3,4-b]pyridines. Assiut Univ. J. Chem. 2006, 35, 45–63. [Google Scholar]
- Jairo, Q.; Diana, M.; Braulio, I.; Ridrigo, A.; Manuel, N.; Adolfo, S.; Justo, C.; John, N. Regioselective synthesis of 4,7,8,9-tetrahydro-2H-pyrazolo[3,4-b]quinolin-5(6H)-ones. Mechanism and structural analysis. Tetrahedron 2001, 57, 6947–6953. [Google Scholar]
- Bazgir, A; Khanaposhanti, M.; Sooki, A. One-pot synthesis and antibacterial activities of pyrazolo[4′,3′:5,6]pyrido[2,3-d]pyrimidine-dione derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 5800–5803. [Google Scholar]
- Chebanov, V.A.; Sakhno, Y.I.; Desenk, S.M.; Chernenko, V.N.; Musatov, V.I.; Shishkina, S.V.; Shishkin, O.V.; Kappe, O. Cyclocondensation reactions of 5-aminopyrazoles, pyruvic acids and aldehydes. Multicomponent approaches to pyrazolopyridines and related products. Tetrahedron 2007, 1229–1242. [Google Scholar]
- Shaabani, A.; Seyyedhamez, M.; Maleki, A.; Behnan, M.; Rezazdeh, F. Synthesis of fully substituted pyrazolo[3,4-b]pyridine-5-carboxamide derivatives via a one-pot four-component reaction. Tetrahedron Lett. 2009, 50, 2911–2913. [Google Scholar] [CrossRef]
- Balamurugan, K.; Perumal, S.; Menedez, J.C. New four-component reaction in water: a convergent approach to the metal-free synthesis of spiro[indoline/ acenaphthylene-3,4'-pyrazolo[3,4-b]pyridine derivatives. Tetrahedron 2011, 67, 3201–3208. [Google Scholar] [CrossRef]
- Shawali, A.S.; Elghandour, A.H.; Sayed, A.R. A novel one-pot synthesis of 3-arylazo[1,2,4]triazolo[4,3-a]pyrimidin-5-(1H)-ones. Synth. Commun. 2001, 31, 731–740. [Google Scholar] [CrossRef]
- Bedford, G.R.; Taylor, P.J.; Webb, G.A. 15N-NMR studies of guanidines. II—The fused-in guanidine unit of some oxoheterocycles: A combined 15N-NMR, 13C-NMR and IR study. Magn. Res. Chem. 1995, 33, 389–394. [Google Scholar]
- Elguero, J.; Goya, P.; Martinez, A.; Rozas, I. On the Tautomerism of 2-Phenacyl-4-pyrimidinones and Related Compounds. Chem. Ber. 1989, 122, 919–924. [Google Scholar] [CrossRef]
- Greenhill, J.V.; Ismail, M.J.; Bedford, G.R.; Edwards, P.N.; Taylor, P.J. Conformational and tautmeric studies of acyl guanidines Part 2. Vibrations and C-13 nuclear magnetic resonance spectroscopy. J. Chem. Soc. Perkin Trans. 1985, 2, 1265–1274. [Google Scholar] [CrossRef]
- Reiter, J.; Bongo, L.; Dyortsok, P. On triazoles XI. structure elucidation of isomeric 1,2,4-triazolopyrimidinones. Tetrahedron 1987, 43, 2497–2504. [Google Scholar]
- Rami, V.J. One-Pot Synthesis of Mono- and Dinitro-1,2,4-triazino[3,2-b]benzothiazoles. Liebigs Ann. Chem. 1988, 11, 1089–1090. [Google Scholar] [CrossRef]
- Heinisch, G.; Holzer, W. Pyrazoles 3. N-1 Protected 4-Substituted Pyrazoles—Synthesis and Nmr Investigation. Heterocycles 1988, 27, 2443–2457. [Google Scholar]
- Khalil, Z.H.; Geies, A.A. Synthesis and reactions of some thieno[2,3-d]pyrimidine derivatives. Phosphorus Sulfur Silicon 1991, 60, 223–231. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Bennett, J.W. Principles of antifungal and antibacterial therapy. Med. Mycol. Lea Febiger. Philadel. 1992, 81–102. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | Diameter of the inihibition zone (mm) MIC (mM) | |||||
|---|---|---|---|---|---|---|
| S. aureus AUMC B.54 | S. cereus AUMC B.52 | M. luteus AUMC B.112 | E. coli AUMC B.53 | p. aeruginosa AUMC B.73 | S. marcescens AUMC B.55 | |
| 6 | - | - | - | - | - | - |
| 7a | - | - | - | - | - | - |
| 9 | - | - | - | - | - | - |
| 11 | 8 (2.5) | - | - | - | - | - |
| 12 | - | 10 (1.25) | 18 (20) | - | - | - |
| 13b | - | - | - | - | - | - |
| 17 | - | - | - | - | - | - |
| 19c | 8 (1.25 ) | 10 (0.15) | 10 (5) | - | - | - |
| 20 | - | - | - | - | - | - |
| 22a | 8 (5) | - | - | - | 11 (20) | - |
| CHL | 10 (0.08) | 12 (1.25) | 12 (2.5) | 10 (0.08) | 12 (0.3) | 13 (1.25) |
| Compound No. | Diameter of the inihibition zone (mm) MIC (mM) | |||||
|---|---|---|---|---|---|---|
| C. albicans AUMC 418 | G.candidum AUMC 226 | T. rubrum AUMC 1804 | A.flavus AUMC 3214 | F. oxysporum AUMC 5119 | S. brevicaylis AUMC 729 | |
| 6 | - | - | - | - | - | - |
| 7a | - | - | - | - | - | - |
| 9 | - | - | - | - | - | - |
| 11 | - | 11 (20) | 8 (20) | - | - | 10 (20) |
| 12 | 9 (2.5) | 15 (5) | 10 (10) | 10 (2.5) | 16 (20) | 20 (5) |
| 13b | - | - | - | - | - | - |
| 17 | 10 (20) | - | 24p.i (20) | 10 (20) | 8 (20) | 12 (10) |
| 19c | 9 (2.5) | 16 (2.5) | 12 (5) | 14 (2.5) | 10 (10) | 28 (5) |
| 20 | - | - | - | - | - | - |
| 22a | 10 (0.6) | - | - | - | - | - |
| CLO | 12 (0.08) | 14 (0.08) | 35 (0.08) | 15 (0.15) | 14 (0.15) | 24 p.i (0.3) |
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
El-Emary, T.I.; El-Mohsen, S.A.A. Multi-Component One-Pot Synthesis and Antimicrobial Activities of 3-Methyl-1,4-diphenyl-7-thioxo-4,6,8,9-tetrahydro-pyrazolo[5,4-b]pyrimidino[5,4-e]pyridine-5-one and Related Derivatives. Molecules 2012, 17, 14464-14483. https://doi.org/10.3390/molecules171214464
El-Emary TI, El-Mohsen SAA. Multi-Component One-Pot Synthesis and Antimicrobial Activities of 3-Methyl-1,4-diphenyl-7-thioxo-4,6,8,9-tetrahydro-pyrazolo[5,4-b]pyrimidino[5,4-e]pyridine-5-one and Related Derivatives. Molecules. 2012; 17(12):14464-14483. https://doi.org/10.3390/molecules171214464
Chicago/Turabian StyleEl-Emary, Talaat I., and Shawkat A. Abd El-Mohsen. 2012. "Multi-Component One-Pot Synthesis and Antimicrobial Activities of 3-Methyl-1,4-diphenyl-7-thioxo-4,6,8,9-tetrahydro-pyrazolo[5,4-b]pyrimidino[5,4-e]pyridine-5-one and Related Derivatives" Molecules 17, no. 12: 14464-14483. https://doi.org/10.3390/molecules171214464
APA StyleEl-Emary, T. I., & El-Mohsen, S. A. A. (2012). Multi-Component One-Pot Synthesis and Antimicrobial Activities of 3-Methyl-1,4-diphenyl-7-thioxo-4,6,8,9-tetrahydro-pyrazolo[5,4-b]pyrimidino[5,4-e]pyridine-5-one and Related Derivatives. Molecules, 17(12), 14464-14483. https://doi.org/10.3390/molecules171214464