Inhibitory Effects of Enalaprilat on Rat Cardiac Fibroblast Proliferation via ROS/P38MAPK/TGF-β1 Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
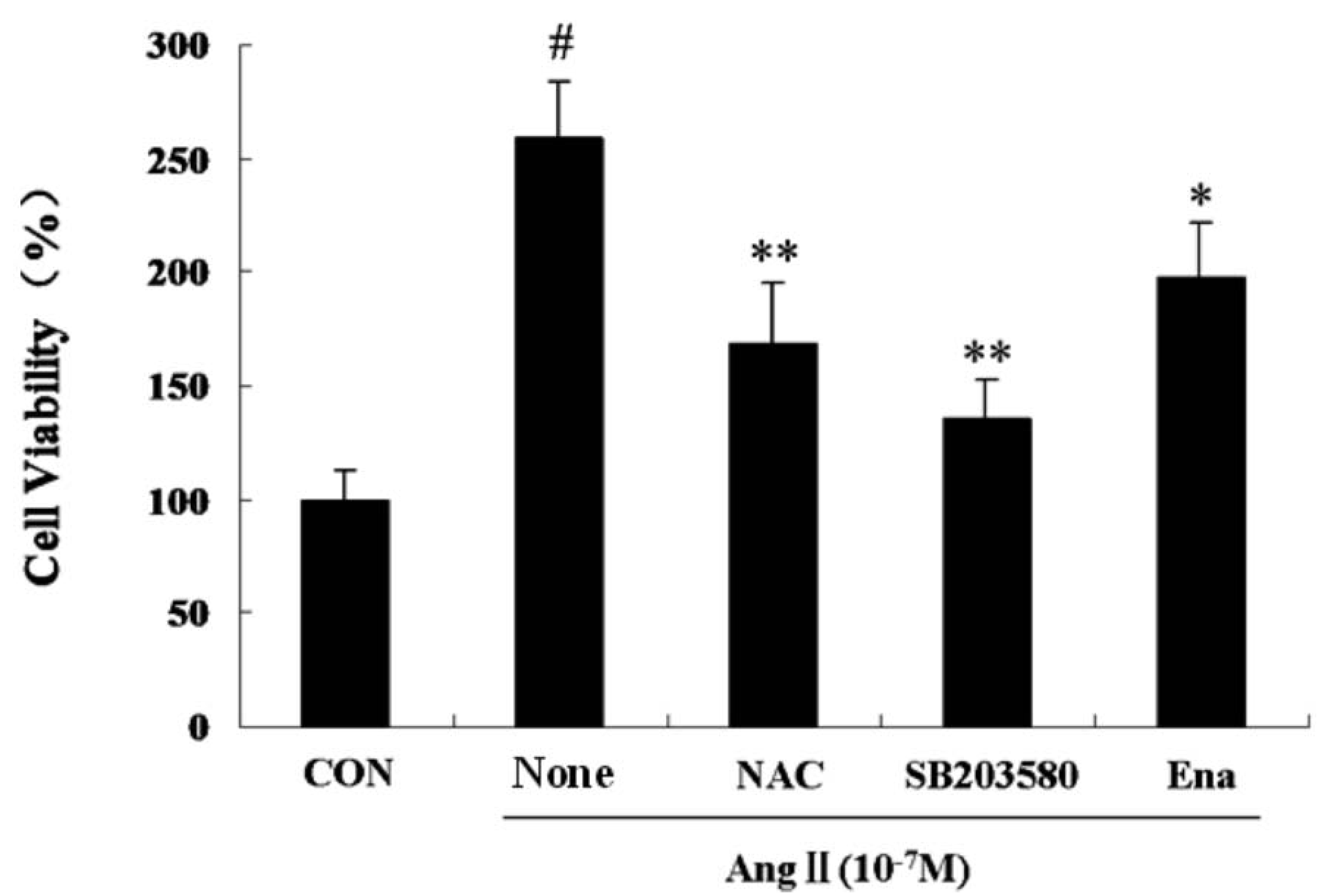
2.1. Effect of Enalaprilat on Cardiac Fibroblast Proliferation

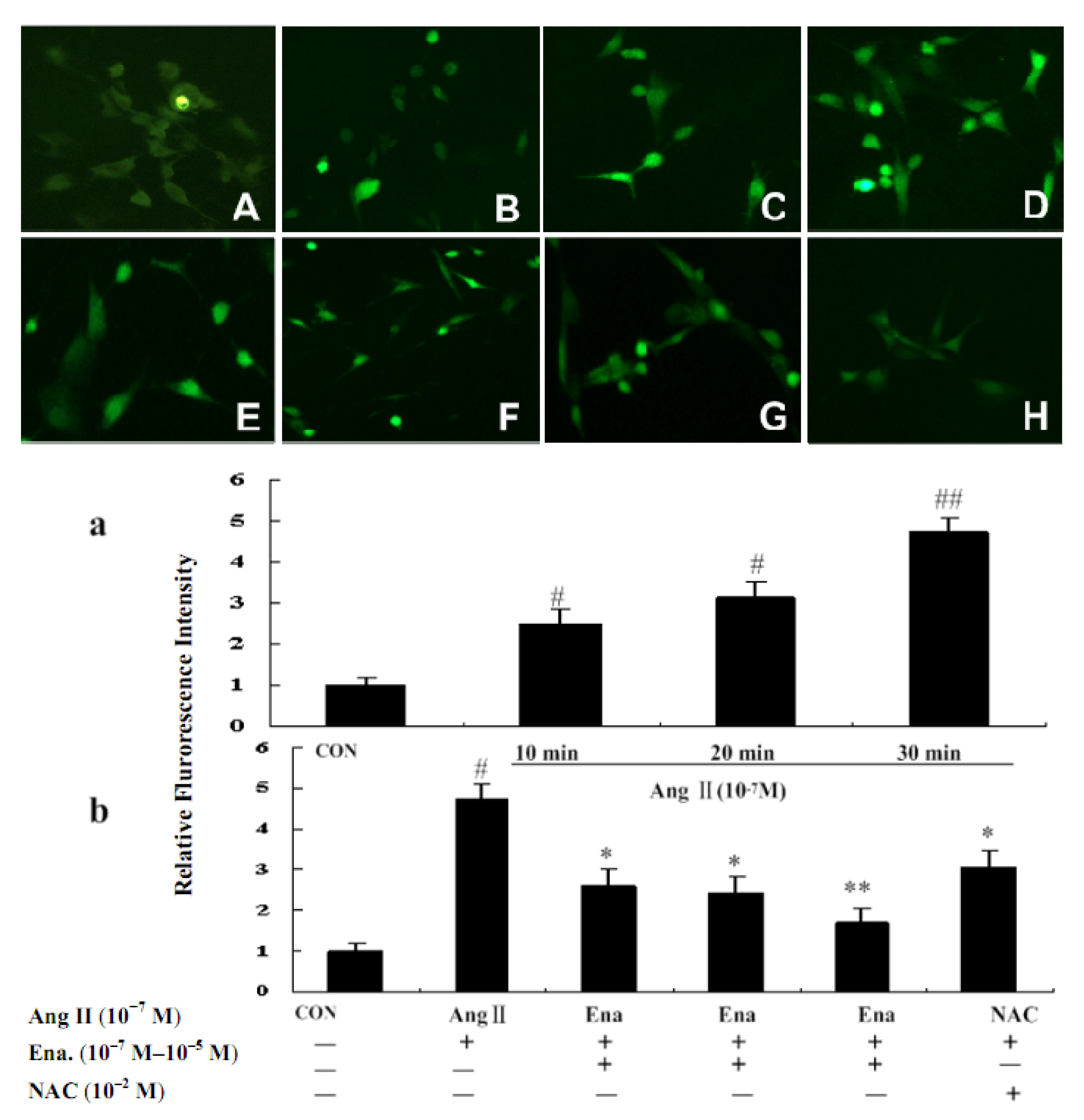
2.2. Effect of Enalaprilat on Intracellular Reactive Oxygen Species Generation

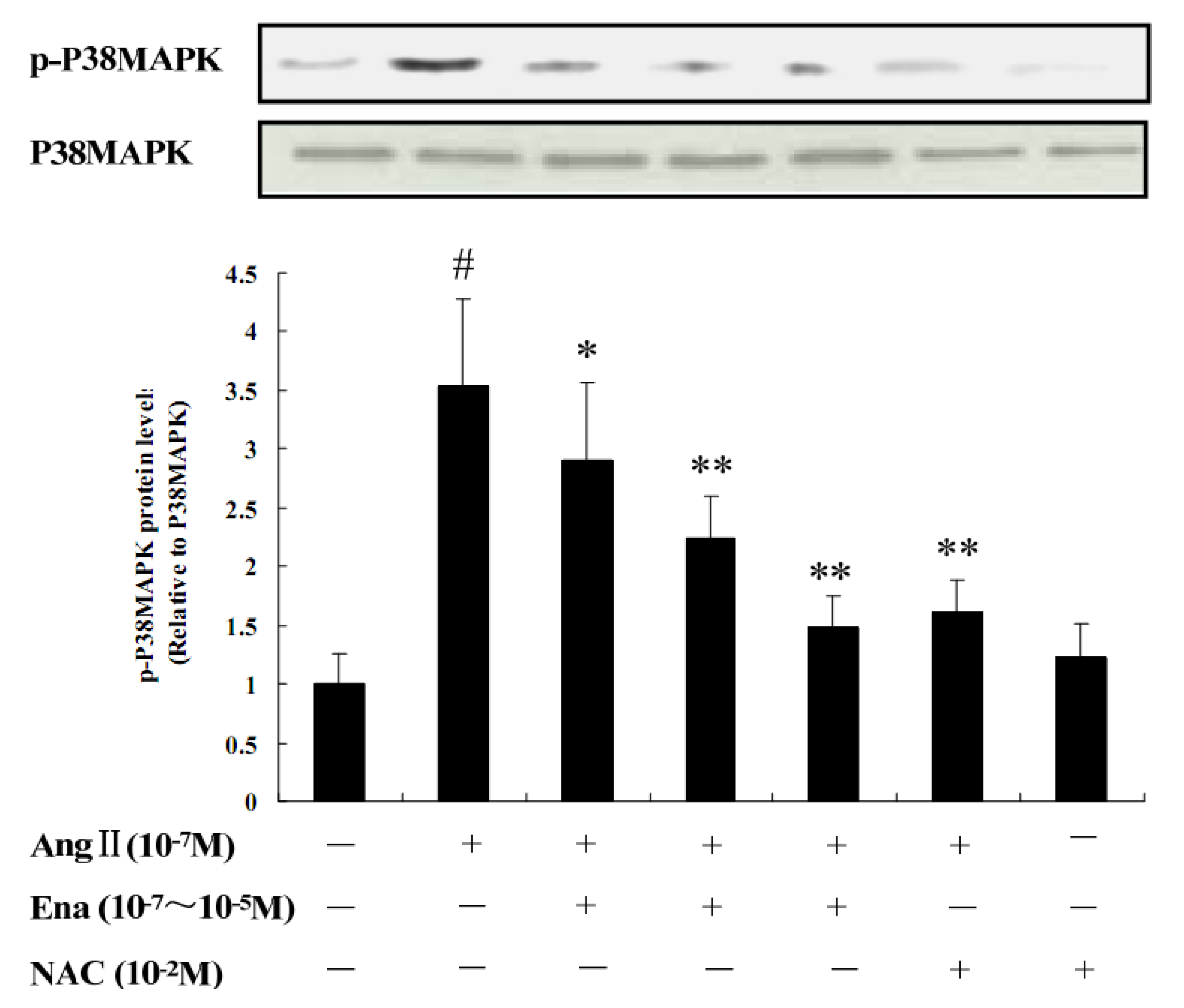
2.3. Protein Analysis of Phosphorylated p38MAPK by Western Blot

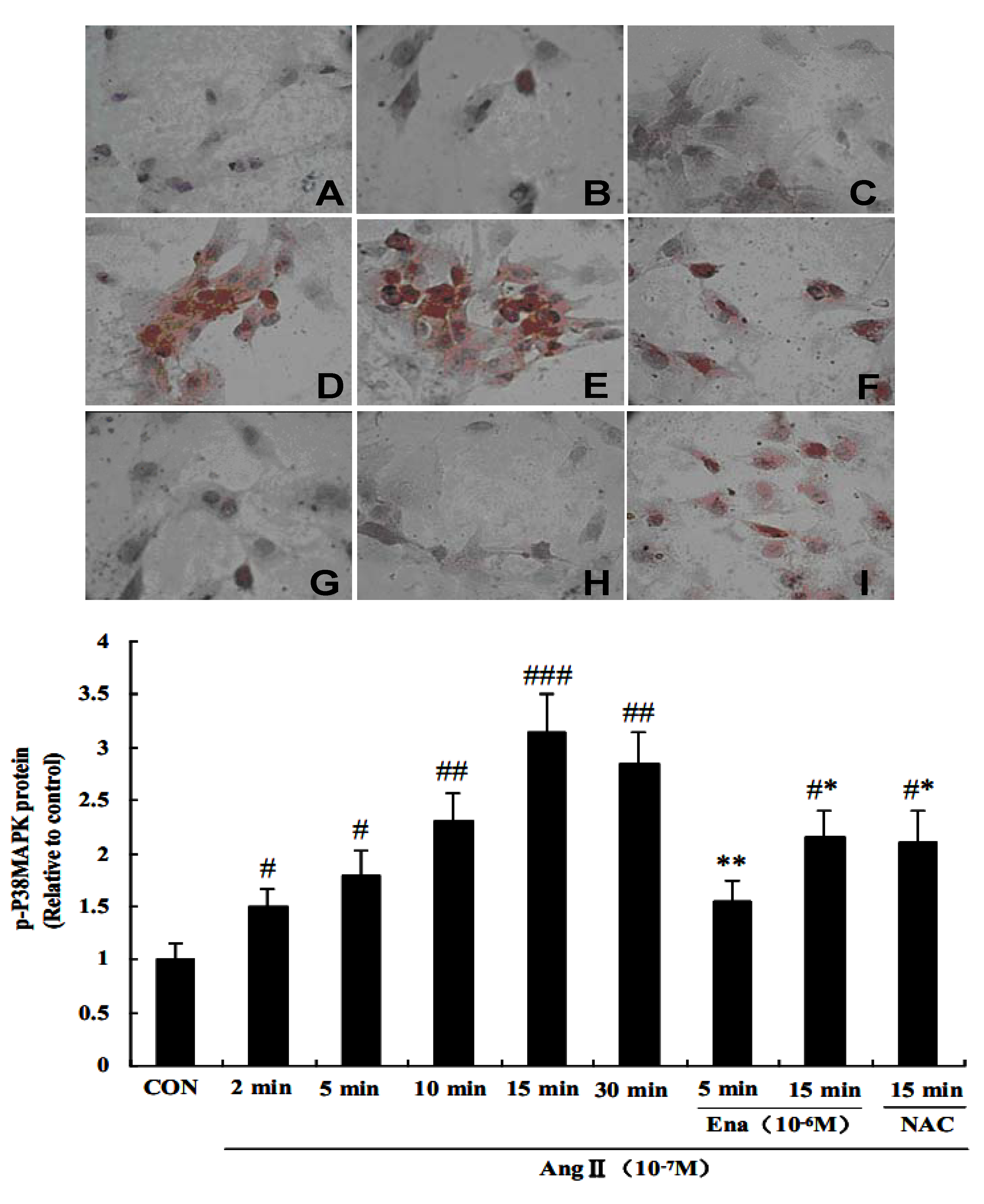
2.4. Effect of Enalaprilat on p38 MAPK Phosphorylation in CFb

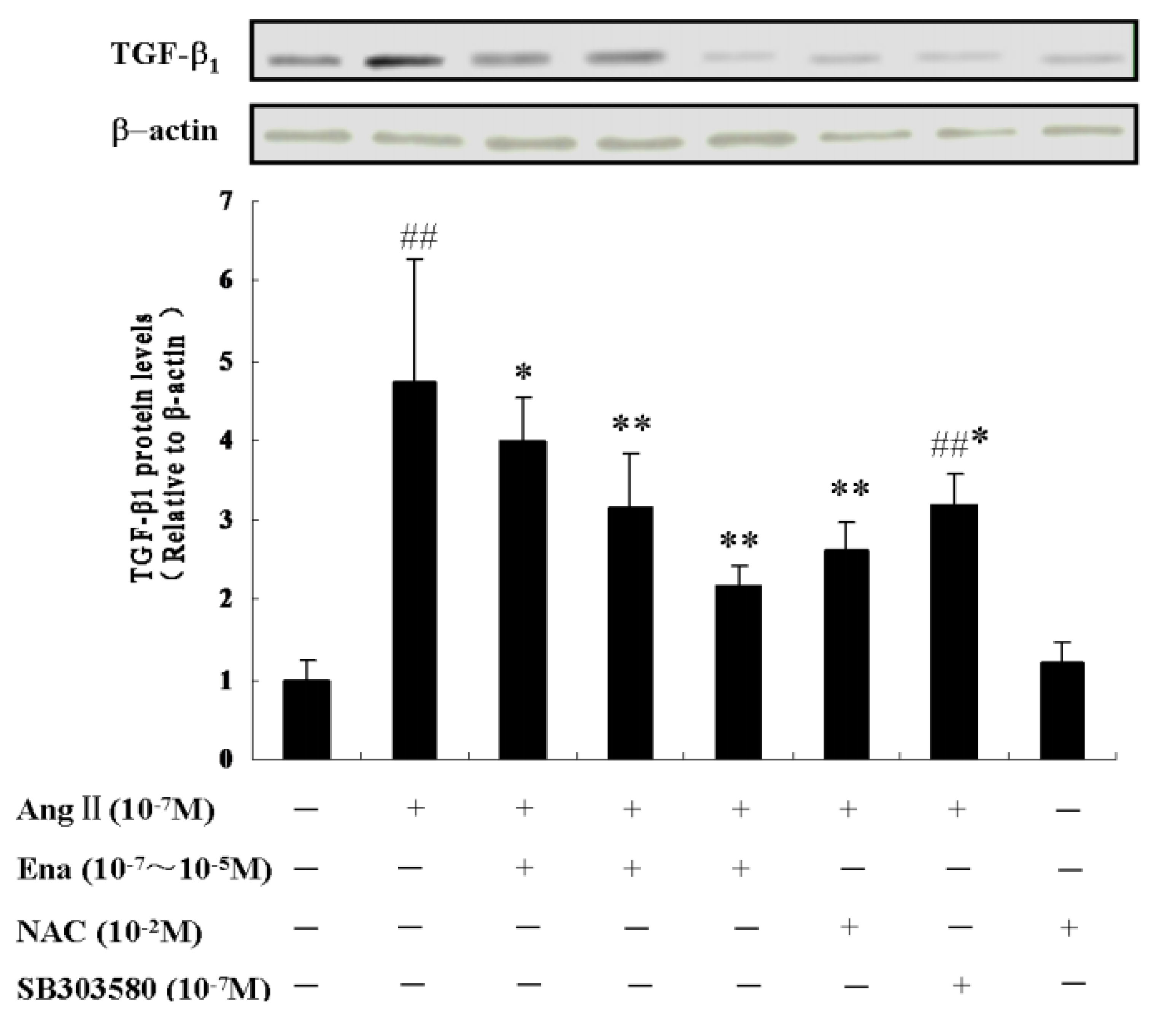
2.5. Effect of Enalaprilat on TGF-β1 Protein Expression

3. Experimental
3.1. Chemicals
3.2. Cell Isolation
3.3. Proliferation Assay
3.4. Immunofluorescence Assay of Intracellular ROS
3.5. Immunocytochemistry
3.6. Western Blot Assay
3.7. Statistical Analysis
4. Conclusion
Acknowledgments
- Samples Availability: Samples are available from the authors.
References and Notes
- Singh, B.M.; Mehta, J.L. Interactions between the renin-angiotensin system and dyslipidemia: Relevance in the therapy of hypertension and coronary heart disease. Arch. Intern. Med. 2003, 163, 1291–1304. [Google Scholar]
- Fujii, T.; Onohara, N.; Maruyama, Y.; Tanabe, S.; Kobayashi, H.; Fukutomi, M.; Nagamatsu, Y.; Nishihara, N.; Inoue, R.; Sumimoto, H.; Shibasaki, F.; Nagao, T.; Nishida, M.; Kurose, H. Gα12/13-mediated Production of Reactive Oxygen Species Is Critical for Angiotensin Receptor-induced NFAT Activation in Cardiac Fibroblasts. J. Boil. Chem. 2005, 17, 23041–23047. [Google Scholar]
- Wang, L.; Li, Y. Dynamic expression of p38MAPK and TGF-β1 in the tubulo-interstitium of diabetic rats. J. North Chin. Coal Med. Coll. 2007, 9, 281–291. [Google Scholar]
- Chin, B.K.; Mohsenin, A.; Li, S.X.; Choi, A.M.; Choi, M.E. Stimulation of pro-α1 (I) colllagen by TGF-β1 in mesangial cells: Role of the p38MAPK pathway. Am. J. Physiol. 2001, 280, F495–F504. [Google Scholar]
- Gruden, G.; Zonca, S.; Hayward, A.; Thomas, S.; Maestrini, S.; Gnudi, L.; Viberti, G.C. Mechanical stretch-induced fibronectin and transforming growth factor -β1 production in human mesangial cells is p38 mitogen-activated protein kinase depedent. Diabetes 2000, 49, 655–661. [Google Scholar]
- Du, N.L.; Qi, W.H.; Qiu, X.S. The Effects of Enalapril and Losartan on Left Ventricular Hypertrophy and Myocardial Fibrosis in SHR. Chin. J. Hypertens. 2000, 8, 261–267. [Google Scholar]
- Sano, M.; Fukuda, K.; Sato, T.; Kawaguchi, H.; Suematsu, M.; Matsuda, S.; Koyasu, S.; Matsui, H.; Yamauchi-Takihara, K.; Harada, M.; Saito, Y.; Ogawa, S. ERK and p38 MAPK, but not NF-κB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ. Res. 2001, 89, 661–669. [Google Scholar] [CrossRef]
- Reckelhoff, J.F.; Romero, J.C. Role of oxidative stress in angiotensin-induced hypertension. Am. J. Physiol. 2003, 284, R893–R912. [Google Scholar]
- Harrison, D.G.; Cai, H.; Landmesser, U.; Griendling, K.K. Interactions of angiotensin II with NAD (P) H oxidase, oxidative stress and cardiovascular disease. J. Renin.-Angio.-Aldo. S. 2003, 4, 51–61. [Google Scholar]
- Engberding, N.; Spiekermann, S.; Schaefer, A.; Heineke, A.; Wiencke, A.; Müller, M.; Fuchs, M.; Hilfiker-Kleiner, D.; Hornig, B.; Drexler, H.; Landmesser, U. Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: A new action for an old drug. Circulation 2004, 110, 2171–2179. [Google Scholar]
- Kim, B.Y.; Han, M.J.; Chung, A.S. Effects of reactive oxygen species on proliferation of Chinese Hamster lung fibroblast (V79) cells. Free Radical Biol. Med. 2001, 30, 686–698. [Google Scholar] [CrossRef]
- Shen, W.L.; Gao, P.J.; Che, Z.Q.; Ji, K.D.; Yin, M.; Yan, C.; Berk, B.C.; Zhu, D.L. NAD(P)H oxidase-derived reactive oxygen species regulate angiotensin-II induced adventitial fibroblast phenotypic differentiation. Biochem. Biophys. Res. Commun. 2006, 339, 331–343. [Google Scholar] [CrossRef]
- See, F.; Thomas, W.; Way, K.; Tzanidis, A.; Kompa, A.; Lewis, D.; Itescu, S.; Krum, H. p38 Mitogen- Activated Protein Kinase Inhibition Improves Cardiac Function and Attenuates Left Ventricular Remodeling Following Myocardial Infarction in the Rat. J. Am. Coll. Cardiol. 2004, 44, 1671–1689. [Google Scholar]
- Takemoto, M.; Node, K.; Nakagami, H.; Liao, Y.; Grimm, M.; Takemoto, Y.; Kitakaze, M.; Liao, J.K. Statins as antioxidative therapy for preventing cardiac myocyte hypertrophy. J. Clin. Invest. 2001, 108, 1421–1438. [Google Scholar]
- Li, D.; Saldeen, T.; Romeo, F.; Mehta, J.L. Oxidized LDL upregulates angiotensin II type 1 receptor expression in cultured human coronary artery endothelial cells: The potential role of transcription factor NF-κB. Circulation 2000, 102, 1971–1976. [Google Scholar]
- Ge, B.; Gram, H.; Di Padova, F.; Huang, B.; New, L.; Ulevitch, R.J.; Luo, Y.; Han, J. MAPKK-independent activation of p38 alpha mediated by TAB 1-dependent autophosporylation of p38 alpha. Science 2002, 295, 1291–1294. [Google Scholar]
- Gruden, G.; Zonca, S.; Hayward, A.; Thomas, S.; Maestrini, S.; Gnudi, L.; Viberti, G.C. Mechanical stretch induced fibronectin and transforming growth factor-β1 production in human mesangial cells is p38 mitogen-activated protein kinase dependent. Diabetes 2000, 49, 651–661. [Google Scholar]
- Khalil, N.; Xu, Y.D.; O’Connor, R.; Duronio, V. Proliferation of pulmonary interstitial fibroblasts is mediated by transforming growth factor-1-induced release of extracellular fibroblast growth factor-2 and phosphorylation of p38 MAPK and JNK. J. Biol. Chem. 2005, 280, 43001–43009. [Google Scholar]
- Suzuki, H.; Uchida, K.; Nitta, K.; Nihei, H. Role of mitogen-activeted protein kinase in the regulation of transforming growth factor-beta induced fibronectin accumulation in cultured renal interstitial fibroblasts. Clin. Exp. Nephrol. 2004, 8, 181–195. [Google Scholar]
- Geleilete, T.J.; Melo, G.C.; Costa, R.S.; Volpini, R.A.; Soares, T.J.; Coimbra, T.M. Role of myofibroblasts, macrophages, transforming growth factor-beta endothelin, angiotensin II and fibronectin in the progression of tubulointestial nephritis induced by gentamycin. J. Nephrol. 2002, 15, 631–642. [Google Scholar]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Radical Biol. Med. 2000, 28, 461–499. [Google Scholar]
- Masutani, H. Oxidative stress response and signaling in hematological malignancies and HIV infection. Int. J. Hematol. 2000, 71, 21–32. [Google Scholar]
- Kim, S.J.; Angel, P.; Lafyatis, R.; Hattori, K.; Kim, K.Y.; Sporn, M.B.; Karin, M.; Roberts, A.B. Autoinduction of transforming growth factor beta 1 is mediated by the AP-1 complex. Mol. Cell. Biol. 1990, 10, 1492–1497. [Google Scholar]
- Birchenall-Roberts, M.C.; Ruscetti, F.W.; Kasper, J.; Lee, H.D.; Friedman, R.; Geiser, A.; Sporn, M.B.; Roberts, A.B.; Kim, S.J. Transcriptional regulation of the transforming growth factor β1 promoter by v-src gene products is mediated through the AP-1 complex. Mol. Cell. Biol. 1990, 10, 4971–4983. [Google Scholar]
- Kim, S.J.; Glick, A.; Sporn, M.B.; Roberts, A.B. Characterization of the promoter region of the human transforming growth factor-β1 gene. J. Biol. Chem. 1989, 264, 401–408. [Google Scholar]
- Inagaki, Y.; Truter, S.; Tanaka, S.; di Liberto, M.; Ramirez, F. Overlapping pathways mediate the opposing actions of tumor necrosis factor-alpha and transforming growth factor-beta on alpha2(I) collagen gene transcription. J. Biol. Chem. 1995, 270, 3353–3358. [Google Scholar]
- Cheng, C.M.; Hong, H.J.; Liu, J.C.; Shih, N.L.; Juan, S.H.; Loh, S.H.; Chan, P.; Chen, J.J.; Cheng, T.H. Crucial Role of Extracellular Signal-Regulated Kinase Pathway in Reactive Oxygen Species-Mediated Endothelin-1 Gene Expression Induced by Endothelin-1 in Rat Cardiac Fibroblasts. Mol. Pharmacol. 2003, 63, 1001–1011. [Google Scholar]
- van Eickels, M.; Grohé, C.; Löbbert, K.; Stimpel, M.; Vetter, H. Angiotensin converting enzyme inhibitors block mitogenic signaling pathways in rat cardiac fibroblasts. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 359, 391–399. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, M.; Zheng, Y.; Sun, H.-X.; Yu, D.-J. Inhibitory Effects of Enalaprilat on Rat Cardiac Fibroblast Proliferation via ROS/P38MAPK/TGF-β1 Signaling Pathway. Molecules 2012, 17, 2738-2751. https://doi.org/10.3390/molecules17032738
Yu M, Zheng Y, Sun H-X, Yu D-J. Inhibitory Effects of Enalaprilat on Rat Cardiac Fibroblast Proliferation via ROS/P38MAPK/TGF-β1 Signaling Pathway. Molecules. 2012; 17(3):2738-2751. https://doi.org/10.3390/molecules17032738
Chicago/Turabian StyleYu, Min, Yang Zheng, Hong-Xia Sun, and Du-Juan Yu. 2012. "Inhibitory Effects of Enalaprilat on Rat Cardiac Fibroblast Proliferation via ROS/P38MAPK/TGF-β1 Signaling Pathway" Molecules 17, no. 3: 2738-2751. https://doi.org/10.3390/molecules17032738
APA StyleYu, M., Zheng, Y., Sun, H.-X., & Yu, D.-J. (2012). Inhibitory Effects of Enalaprilat on Rat Cardiac Fibroblast Proliferation via ROS/P38MAPK/TGF-β1 Signaling Pathway. Molecules, 17(3), 2738-2751. https://doi.org/10.3390/molecules17032738